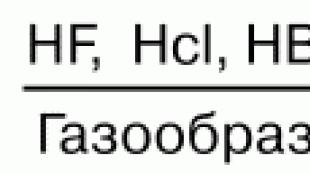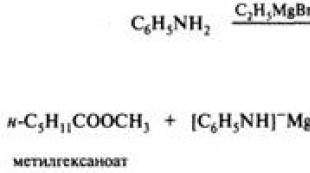Üks estrite hüdrolüüsi saadusi on. Estrite hüdrolüüs
Estrid on karboksüülhapete funktsionaalsed derivaadid üldvalemiga RC(0)0R."
Omandamise meetodid. Kõige olulisem viis estrite saamiseks on alkoholide ja fenoolide atsüülimine erinevate atsüülivate ainetega, näiteks karboksüülhape, happekloriidid, anhüdriidid. Neid võib saada ka Tištšenko reaktsiooniga.
Estreid valmistatakse suure saagisega karboksüülhappe soolade alküülimisel alküülhalogeniididega:
Estrid tekivad karboksüülhapete elektrofiilsel lisamisel alkeenidele ja alküünidele. Reaktsiooni kasutatakse sageli tertsiaarsete alkoholiestrite, nt. hõõrub-butüüleetrid:
Äädikhappe lisamisel atsetüleenile tekib tööstuslikult oluline monomeer vinüülatsetaat, Katalüsaatorina kasutatakse tsinkatsetaati aktiivsöel:
Hüdrolüüs. Atsüülimisreaktsioonidest kõige olulisem on estrite hüdrolüüs alkoholi ja karboksüülhappe moodustumisega:
Reaktsioon toimub nii happelises kui aluselises keskkonnas. Estrite happega katalüüsitud hüdrolüüs - esterdamise pöördreaktsioon, toimub sama mehhanismi järgi Als 2
Leeliseline hüdrolüüs on pöördumatu, reaktsiooni käigus kulub eetri mooli kohta üks mool leelist, st leelis toimib selles reaktsioonis tarbitava reaktiivina, mitte katalüsaatorina:
Estrite hüdrolüüs leeliselises keskkonnas toimub bimolekulaarse atsüülmehhanismi kaudu VAS2 läbi tetraeedrilise vaheühendi (I) moodustumise etapi. Aluselise hüdrolüüsi pöördumatuse tagab karboksüülhappe (I) ja alkoksiidiooni (III) praktiliselt pöördumatu happe-aluse interaktsioon. Saadud karboksüülhappe anioon (IV) on ise üsna tugev nukleofiil ja seetõttu ei allu nukleofiilsele rünnakule.
Ümberesterdamine. Selle reaktsiooni abil viiakse sama happe estrite vastastikune muundamine läbi vastavalt järgmisele skeemile:
Ümberesterdamine on pöörduv protsess, mida katalüüsivad nii happed kui ka alused ja mis toimub samade mehhanismide järgi nagu estrite esterdamise ja hüdrolüüsi reaktsioonid. Tasakaalu nihutatakse tuntud meetoditega, nimelt reaktiivi alkoholi kasutamisega (ülaloleval diagrammil R"OH - paremale nihutamiseks) või ühe reaktsiooniprodukti destilleerimisega, kui see on madalaima keemistemperatuuriga. Näiteks ümberesterdamise teel saadakse hästi tuntud anesteetikum novokaiin(alus) l-aminobensoehappe etüülestrist:
Estri kondensatsioon. Kui kaks estri molekuli kondenseeruvad aluselise katalüsaatori juuresolekul, tekivad β-oksohappe estrid:
Etüülatsetaadi molekulil on esterrühma induktiivse toime tõttu nõrgad CH-happe omadused ja see on võimeline interakteeruma tugeva alusega - etoksiidiooniga:
Karboksüülhapete amiidid. Omandamise meetodid. Amiidrühma struktuur. Amiidide happe-aluselised omadused. Happeline ja aluseline hüdrolüüs. Amiidide lõhustamine halogeenidega leeliselises keskkonnas ja lämmastikhappes. Dehüdratsioon nitriilideks.
Amiidid on karboksüülhapete funktsionaalsed derivaadid üldvalemiga R-C(O)-NH2_nR"", kus P = 0-2.
Omandamise meetodid. Kõige olulisem meetod amiidide valmistamiseks on ammoniaagi ja amiinide atsüülimine happehalogeniidide, anhüdriidide ja estritega.
Ammoniaagi ja amiinide atsüülimine happehalogeniididega. Ammoniaagi ja amiinide atsüülimisreaktsioon happehalogeniididega on eksotermiline ja toimub jahutamisel:
Ammoniaagi ja amiinide atsüülimine anhüdriididega. Amiinide atsetüülimiseks kasutatakse kõige sagedamini kõige kättesaadavamat anhüdriidi, atseetanhüdriidi:
Estrite ammonolüüs. Amiide saadakse estrite ammonolüüsi teel. Näiteks kui ammoniaagi vesilahus reageerib dietüülfumaraadiga, moodustub täielik fumaarhappeamiid:
Amiidide struktuur. Amiidrühma elektrooniline struktuur on suures osas sarnane karboksüülrühma struktuuriga. Amiidrühm on p,l-konjugeeritud süsteem, milles lämmastikuaatomi üksik elektronide paar on konjugeeritud C=0 sideme elektronidega. Amiidrühma elektrontiheduse delokaliseerimist saab kujutada kahe resonantsstruktuuriga:
Konjugatsiooni tõttu on C-N side amiidides osaliselt topeltseotud, selle pikkus on oluliselt väiksem kui amiinide üksiksideme pikkus, samas kui C=0 side on veidi pikem kui C=0 side aldehüüdides ja ketoonides. Amiidrühmal on konjugatsiooni tõttu lame konfiguratsioon. Allpool on toodud iV-asendatud amiidi molekuli geomeetrilised parameetrid, mis on määratud röntgendifraktsioonianalüüsi abil:
Happe-aluse omadused. Amiididel on nõrgad happelised ja aluselised omadused. Amiidide aluselisus jääb pA"in+ väärtuste vahemikku -0,3 kuni -3,5. Amiidide aminorühma aluselisuse vähenemise põhjuseks on lämmastikuaatomi üksiku elektronpaari konjugatsioon karbonüülrühmaga. Tugevate hapetega interakteerudes protoneerivad amiidid hapnikuaatomi juures nagu hapete lahjendatud ja kontsentreeritud lahustes. Selline interaktsioon on happelise katalüüsi aluseks amiidide hüdrolüüsireaktsioonides:
Atsüülimisreaktsioonid. Tugeva elektrone loovutava aminorühma olemasolu tõttu konjugeeritud amiidsüsteemis on karbonüüli süsinikuaatomi elektrofiilsus ja seega ka amiidide reaktsioonivõime atsüülimisreaktsioonides väga madal. Amiidide madal atsüülimisvõime on seletatav ka sellega, et amiidioon NH2- on halb lahkuv rühm. Atsüülimisreaktsioonidest on praktilise tähtsusega amiidide hüdrolüüs, mida saab läbi viia happelises ja aluselises keskkonnas. Amiide on palju raskem hüdrolüüsida kui teisi karboksüülhapete funktsionaalseid derivaate. Amiidide hüdrolüüs toimub estrite hüdrolüüsiga võrreldes rangemates tingimustes.
Amiidide happeline hüdrolüüs on pöördumatu reaktsioon, mis põhjustab karboksüülhappe ja ammooniumisoola moodustumist:
Aluseline hüdrolüüs on samuti pöördumatu reaktsioon; selle tulemusena moodustuvad karboksüülhappe sool ja ammoniaak või amiin:
Lämmastikhappe seedimine. Lämmastikhappe ja teiste nitroseerivate ainetega koostoimel muundatakse amiidid vastavateks karboksüülhapeteks saagisega kuni 90%.
Süsinikhape ja selle funktsionaalsed derivaadid; fosgeen, klorosüsiniku eetrid, karbaamhape ja selle estrid (uretaanid). Uurea (uurea), aluselised ja nukleofiilsed omadused. Karbamiidi hüdrolüüs. Atsüüluuread (ureiidid), ureiidhapped. Karbamiidi koostoime lämmastikhappe ja hüpobromiitidega. Guanidiin, põhiomadused.
Süsinikhape ei kuulu traditsiooniliselt orgaaniliste ühendite hulka, kuid tal endal ja selle funktsionaalsetel derivaatidel on teatav sarnasus karboksüülhapete ja nende derivaatidega ning seetõttu käsitletakse neid selles peatükis.
Kahealuseline süsihape on ebastabiilne ühend, mis laguneb kergesti süsinikdioksiidiks ja veeks. Süsinikdioksiidi vesilahuses on süsihappe kujul ainult 0,1%. Süsinikhape moodustab kaks funktsionaalsete derivaatide seeriat – täielikud (keskmised) ja mittetäielikud (happelised). Happe estrid, amiidid ja muud derivaadid on ebastabiilsed ja lagunevad süsinikdioksiidi vabanemiseks:
Täielik süsihappekloriid - fosgeen COC1 2 - on madala keemistemperatuuriga vedelik mädaheina lõhnaga, väga mürgine, põhjustab kopsuturset ja tekib kahjuliku lisandina kloroformi fotokeemilise oksüdatsiooni käigus, mis on tingitud kloroformi ebaõigest säilitamisest. viimane.
Tööstuses toodetakse fosgeeni süsinik(II)monooksiidi radikaalse kloorimise teel aktiivsöega täidetud reaktoris:
Fosgeenil, nagu ka karboksüülhapete happekloriididel, on kõrge atsüülimisvõime, sellest saadakse palju teisi süsihappe funktsionaalseid derivaate.
Fosgeeni reageerimisel alkoholidega moodustuvad kahte tüüpi estrid - täielikud (karbonaadid) ja osalised (süsinikkloriidestrid ehk kloroformaadid), viimased on nii estrid kui ka happekloriidid. Vesinikkloriidi aktseptorina ja nukleofiilse katalüsaatorina kasutatakse tertsiaarseid amiine või püridiini.
Karbaamhape- mittetäielik süsihappeamiid - ebastabiilne ühend, laguneb ammoniaagiks ja süsinikdioksiidiks:
Karbaamhappe estrid - karbamaadid, või uretaanid, - stabiilsed ühendid, mis saadakse alkoholide lisamisel isotsüanaatidele või ammoniaagi ja amiinide atsüülimisel vastava kloroformaadiga:
Uurea(karbamiid) - süsihappe täielik amiid - eraldas esmakordselt uriinist I. Ruel (1773). See on imetajate valkude metabolismi kõige olulisem lõpp-produkt; täiskasvanud inimene eritab päevas 25-30 g karbamiidi. Karbamiidi sünteesis esmakordselt F. Wöhler (1828) ammooniumtsüanaadi kuumutamisel:
See süntees oli esimene näide orgaanilise aine saamisest anorgaanilisest ühendist.
Tööstuses toodetakse uureat ammoniaagist ja süsinikdioksiidist kõrgendatud rõhul ja temperatuuril (180-230 °C, 150-200 atm):
Karbamiid on nõrkade aluseliste omadustega (p.iHvn + 0,1) ja moodustab tugevate hapetega sooli. Lämmastik- ja oksaalhappe soolad on vees lahustumatud.
Karbamiid protoneeritakse pigem hapnikuaatomi kui lämmastikuaatomi juures. Tõenäoliselt on see tingitud lämmastikuaatomite üksikute elektronpaaride delokaliseerumisest p,π konjugatsiooni tõttu.
Keevas vees hüdrolüüsub karbamiid, moodustades ammoniaagi ja süsinikdioksiidi; happed ja alused katalüüsivad seda reaktsiooni:
Karbamiidi kuumutamisel moodustuvad peamised tooted on ammoniaak ja isotsüaanhape. Isotsüaanhape võib trimeriseeruda tsüanuurhappeks või kondenseeruda teise uureamolekuliga, moodustades biureedi. Sõltuvalt kuumutamiskiirusest domineerib üks või teine uurea lagunemise tee:
Hüpohaliidide toime põhjustab ka uurea lagunemist. Olenevalt tingimustest võib moodustuda lämmastik või hüdrasiin; Täpselt nii saadakse viimast tööstuses:
Karbamiidil on ka nukleofiilsed omadused alküülimis- ja atsüülimisreaktsioonides. Karbamiidi alküülimine, olenevalt alküülivast ainest, võib viia O- ja TV-alküülderivaatideni:
Guanidiini ehk iminouureat (H 2 N) 2 C=NH toodetakse tööstuslikult uurea sulatamisel ammooniumnitraadiga või ortokarbonhappe estrite kuumutamisel ammoniaagiga:
Guanidiin on tugevate aluseliste omadustega värvitu kristalne aine. Kõrge aluselisus leelismetallide hüdroksiidide tasemel on tingitud positiivse laengu täielikust delokaliseerumisest sümmeetrilises guanidiiniumi katioonis:
Guanidiini ja biguanidiini jääke leidub mõnedes looduslikes ühendites ja raviainetes.
Estrid nimetatakse üldvalemiga karboksüülhapete funktsionaalseteks derivaatideks RC(O)VÕI" .
Karboksüülhapete (nagu ka sulfoonhapete) estreid nimetatakse sarnaselt sooladele, ainult katiooni nimetuse asemel kasutatakse vastava alküüli või arüüli nimetust, mis asetatakse aniooni nime ette ja kirjutatakse koos seda. Estrirühma -COOR olemasolu võib kajastada ka kirjeldavalt, näiteks "(sellise ja sellise) happe R-ester" (see meetod on oma kohmakuse tõttu vähem eelistatav):
Madalamate alkoholide ja karboksüülhapete estrid on meeldiva lõhnaga lenduvad vedelikud, mis lahustuvad vees halvasti ja lahustuvad hästi enamikes orgaanilistes lahustites. Estrite lõhnad meenutavad erinevate puuviljade lõhnu, mistõttu toiduainetööstuses valmistatakse neid puuviljalõhna imiteerivate essentside valmistamiseks. Estrite suurenenud lenduvust kasutatakse analüütilistel eesmärkidel.
Hüdrolüüs. Atsüülimisreaktsioonidest kõige olulisem on estrite hüdrolüüs alkoholi ja karboksüülhappe moodustumisega:
Reaktsioon toimub nii happelises kui aluselises keskkonnas. Happega katalüüsitud estrite hüdrolüüs - esterdamise pöördreaktsioon toimub sama mehhanismi kohaselt A AC 2:
Selle reaktsiooni nukleofiil on vesi. Tasakaalu nihe alkoholi ja happe moodustumise suunas tagatakse liigse vee lisamisega.
Leeliseline hüdrolüüs on pöördumatu, reaktsiooni käigus kulub eetri mooli kohta üks mool leelist, st leelis toimib selles reaktsioonis tarbitava reagendina, mitte katalüsaatorina:
Estrite hüdrolüüs aluseline keskkond kulgeb bimolekulaarse atsüülmehhanismi B kaudu AC 2 läbi tetraeedrilise vaheühendi (I) moodustumise etapi. Aluselise hüdrolüüsi pöördumatuse tagab karboksüülhappe (II) ja alkoksiidiooni (III) praktiliselt pöördumatu happe-aluse interaktsioon. Saadud karboksüülhappe anioon (IV) on ise üsna tugev nukleofiil ja seetõttu ei allu nukleofiilsele rünnakule.
Estrite ammonolüüs. Amiide saadakse estrite ammonolüüsi teel. Näiteks kui ammoniaagi vesilahus reageerib dietüülfumaraadiga, moodustub täielik fumaarhappeamiid:
Estrite ammonolüüsil madala nukleofiilsusega amiinidega muundatakse viimased kõigepealt leelis- või leelismuldmetallide amiidideks:

Karboksüülhapete amiidid: nomenklatuur; amiidrühma struktuur; happe-aluse omadused; happeline ja aluseline hüdrolüüs; poolitamine hüpobromiitide ja lämmastikhappega; dehüdratsioon nitriilideks; keemiline identifitseerimine.
amiidid nimetatakse üldvalemiga karboksüülhapete funktsionaalseteks derivaatideks R-C(O)-NH2-n R"n, kus n = 0-2. Asendamata amiidides on atsüüljääk ühendatud asendamata aminorühmaga; N-asendatud amiidides on üks vesinikuaatomitest asendatud ühe alküül- või arüülradikaaliga, N,N-asendatud amiidides kahega.
Ühendeid, mis sisaldavad ühte, kahte või kolme lämmastikuaatomiga seotud atsüülrühma, nimetatakse üldiselt amiididena (vastavalt primaarseteks, sekundaarseteks ja tertsiaarseteks). Asendamata rühmaga - NH2 - primaarsete amiidide nimetused on tuletatud vastavate atsüülradikaalide nimedest, asendades järelliide -õli (või -üül) -amiidiga. -karboksüülhappe sufiksiga hapetest moodustunud amiidid saavad järelliite -karboksamiid. Sulfoonhappe amiide nimetatakse ka vastavate hapete järgi, kasutades järelliidet -sulfoonamiid.
RCO-NH- radikaalide nimed (nagu RSO 2 -NH-) moodustatakse amiidide nimedest, muutes järelliide -amiid -amido-. Neid kasutatakse juhul, kui ülejäänud molekul sisaldab kõrgemat rühma või kui asendus toimub keerulisemas struktuuris kui R-radikaal:

N-asendatud primaarsete amiidide RCO-NHR" ja RCO-NR"R" (nagu ka sarnaste sulfoonamiidide) nimetustes on radikaalide R" ja R" nimetused märgitud enne amiidi nimetust sümboliga N. -:
Seda tüüpi amiide nimetatakse sageli sekundaarseteks ja tertsiaarseteks amiidideks, mida IUPAC ei soovita.
N-fenüül-asendatud amiididele on antud nende nimedes järelliide -aniliid. Asendajate asukoht aniliinijäägis on tähistatud numbritega koos algarvudega:
Lisaks on säilinud poolsüstemaatilised nimetused, milles liide -amiid on kombineeritud karboksüülhappe ladinakeelse nimetuse alusega (formamiid, atseetamiid), samuti mõned triviaalne nimetused nagu "aniliidid" (atsüülitud aniliinid) või "toluidiidid" (atsüülitud toluidiinid).
Amiidid on suhteliselt kõrgete ja selgelt eristatavate sulamistemperatuuridega kristalsed ained, mis võimaldab mõnda neist kasutada derivaatidena karboksüülhapete tuvastamiseks. Harvadel juhtudel on need vedelikud, näiteks sipelghappeamiidid - formamiid ja N,N-dimetüülformamiid - tuntud dipolaarsed aprotoonsed lahustid. Madalamad amiidid lahustuvad vees hästi.
Amiidid on üks hüdrolüüsile vastupidavamaid karboksüülhapete funktsionaalsed derivaadid, tänu millele on need looduses laialt levinud. Ravimitena kasutatakse palju amiide. Ligikaudu sajandi jooksul on meditsiinipraktikas kasutatud paratsetamooli ja fenatsetiini, mis on äädikhappe asendatud amiidid.
Amiidide struktuur. Amiidrühma elektrooniline struktuur on suures osas sarnane karboksüülrühma struktuuriga. Amiidrühm on p,π-konjugeeritud süsteem, milles lämmastikuaatomi üksik elektronide paar on konjugeeritud C=O π sideme elektronidega. Amiidrühma elektrontiheduse delokaliseerimist saab kujutada kahe resonantsstruktuuriga:
Konjugatsiooni tõttu on amiidides C-N side osaliselt bilinkitud iseloomu, selle pikkus on oluliselt väiksem kui üksiksideme pikkus amiinides, samas kui C=O side on veidi pikem kui C=O side aldehüüdides ja ketoonides. Amiidrühm konjugatsiooni tõttu on tasase konfiguratsiooniga . Allpool on toodud N-asendatud amiidi molekuli geomeetrilised parameetrid, mis on määratud röntgendifraktsioonianalüüsi abil:

C-N sideme osaliselt topeltseotud olemuse oluliseks tagajärjeks on üsna kõrge energiabarjäär selle sideme ümber pöörlemisel; näiteks dimetüülformamiidi puhul on see 88 kJ/mol. Sel põhjusel võivad amiidid, millel on lämmastikuaatomil erinevad asendajad, esineda π-diastereomeeridena. N-asendatud amiidid eksisteerivad valdavalt Z-isomeeridena:

N,N-diasendatud amiidide puhul sõltub E- ja Z-isomeeride suhe lämmastikuaatomiga seotud radikaalide mahust. Amiidstereoisomeerid on konfiguratsiooniliselt ebastabiilsed, nende olemasolu on tõestatud peamiselt füüsikalis-keemiliste meetoditega, üksikul kujul on neid eraldatud vaid üksikutel juhtudel. Selle põhjuseks on asjaolu, et amiidide pöörlemisbarjäär ei ole endiselt nii kõrge kui alkeenide puhul, mille puhul see on 165 kJ/mol.
Happe-aluse omadused. Amiididel on nõrgad nii happelised kui aluselised omadused . Amiidide aluselisus jääb Pk BH + väärtuste vahemikku -0,3 kuni -3,5. Amiidide aminorühma vähenenud aluselisuse põhjuseks on lämmastikuaatomi üksiku elektronpaari konjugatsioon karbonüülrühmaga. Tugevate hapetega suhtlemisel protoneeritakse amiidid hapnikuaatomi juures nii lahjendatud kui ka kontsentreeritud happelahustes. Selline suhtlus on aluseks happeline katalüüs amiidi hüdrolüüsi reaktsioonides:
Esinevad asendamata ja N-asendatud amiidid nõrgad NH-happe omadused , mis on võrreldav alkoholide happesusega ja eemaldab prootoni ainult reaktsioonides tugevate alustega.
Amiidide moodustumise aluseks on happe-aluse interaktsioonid molekulidevahelised kaaslased , mille olemasolu seletab amiidide kõrget sulamis- ja keemistemperatuuri. Võimalikud on kahte tüüpi assotsieerunud ühendid: lineaarsed polümeerid ja tsüklilised dimeerid. Ühe või teise tüübi ülekaalu määrab amiidi struktuur. Näiteks N-metüülatseetamiid, mille puhul eelistatakse Z-konfiguratsiooni, moodustab lineaarse assotsiatsiooni ja jäigalt fikseeritud E-konfiguratsiooniga laktaamid moodustavad dimeere:

N,N-diasendatud amiidid moodustavad dimeere kahe polaarse molekuli dipool-dipool interaktsiooni tõttu:

Atsüülimisreaktsioonid. Tugeva elektrone loovutava aminorühma olemasolu tõttu konjugeeritud amiidsüsteemis on karbonüüli süsinikuaatomi elektrofiilsus ja seega ka amiidide reaktsioonivõime atsüülimisreaktsioonides väga madal. Amiidide madal atsüülimisvõime on seletatav ka asjaoluga, et amiidioon NH2 - on halb lahkuv rühm. Atsüülimisreaktsioonidest on oluline amiidide hüdrolüüs, mida saab läbi viia happelises ja aluselises keskkonnas. Amiide on palju raskem hüdrolüüsida kui teisi karboksüülhapete funktsionaalseid derivaate. Amiidide hüdrolüüs toimub estrite hüdrolüüsiga võrreldes rangemates tingimustes.
Happeline hüdrolüüs amiidid - pöördumatu reaktsioon, mis põhjustab karboksüülhappe ja ammooniumisoola moodustumist:
Enamikul juhtudel toimub amiidide happeline hüdrolüüs vastavalt mehhanismile bimolekulaarne happe atsüülimine A AC 2 st sarnane estrite happelise hüdrolüüsi mehhanismiga. Reaktsiooni pöördumatus on tingitud asjaolust, et ammoniaak või amiin muundatakse happelises keskkonnas ammooniumiooniks, millel ei ole nukleofiilseid omadusi:

Leeliseline hüdrolüüs Sama pöördumatu reaktsioon; selle tulemusena moodustuvad karboksüülhappe sool ja ammoniaak või amiin:
Amiidide aluseline hüdrolüüs, nagu estrite hüdrolüüs, toimub vastavalt tetraeedriline mehhanism IN AC 2 . Reaktsioon algab hüdroksiidiooni (nukleofiili) lisamisega amiidrühma elektrofiilsele süsinikuaatomile. Saadud anioon (I) protoneeritakse lämmastikuaatomi juures ja seejärel moodustub bipolaarses ioonis (II) hea lahkuv rühm - ammoniaagi või amiini molekul. Arvatakse, et aeglane etapp on tetraeedrilise vaheühendi (II) lagunemine.
Aniliidide ja muude amiidide puhul, mille lämmastikuaatomi juures on elektrone eemaldavad asendajad, võib tetraeedrilise vaheühendi (I) lagunemine toimuda dianiooni (II) moodustumisega:

Lämmastikhappe seedimine. Lämmastikhappe ja teiste nitroseerivate ainetega koostoimel muundatakse amiidid vastavateks karboksüülhapeteks saagisega kuni 90%.
Dehüdratsioon. Fosfor(V)oksiidi ja mõnede teiste reaktiivide (POC1 3, PC1 5, SOCl 2) mõjul asendamata amiidid muudetakse nitriilideks:
![]()
47. Karboksüülhapped: halogeenimine Gell-Volhard-Zelinsky järgi, reaktsiooni kasutamine sünteesiks a -hüdroksü ja a -aminohapped.
Alifaatsete karboksüülhapete halogeenimine.
Alifaatsed karboksüülhapped halogeenitakse katalüütiliste koguste juuresolekul kloori või broomi abil α-positsiooni punane fosfor või fosforhalogeniidid (Gell-Volhard-Zelinsky reaktsioon ). Näiteks kui heksaanhapet broomitakse punase fosfori või fosfor(III)kloriidi juuresolekul, moodustub 2-bromoheksaanhape suure saagisega, näiteks:
Broomimist ei toimu mitte karboksüülhape ise, vaid sellest in situ moodustunud happekloriid. Happekloriidil on tugevamad CH-happe omadused kui karboksüülhappel ja see moodustab kergemini enoolvormi.
Enool (I) lisab broomi, moodustades halogeeni derivaadi (II), mis seejärel elimineerib vesinikhalogeniidi ja muutub α-halogeenitud happehalogeniidiks (III). Viimases etapis regenereeritakse asendamata karboksüülhappe happehalogeniid.

Saadud α-halogeen-asendatud hapetest sünteesitakse nukleofiilsete asendusreaktsioonide abil teisi heterofunktsionaalseid happeid.
Estrite ja kõigi teiste happederivaatide hüdrolüüs nõuab happelist või aluselist katalüüsi. Happelisel hüdrolüüsil tekivad karboksüülhapped ja alkoholid (pöördesterdamise reaktsioon), aluselise hüdrolüüsi käigus tekivad karboksüülhapete ja alkoholide soolad.
Estrite happeline hüdrolüüs:
Mehhanism S N , nukleofiil - H 2 O, alkoksürühm on asendatud hüdroksüülrühmaga.
Estrite leeliseline hüdrolüüs: reaktsioon kulgeb kahes etapis 2 mooli alusega, saadud hape muudetakse soolaks.
Mehhanism S N, Nu = − OH
Soolataoliste ühendite moodustumine Amiidid on neutraalsed ained, kuna ammoniaagi põhiomadusi nõrgestab selles sisalduva vesinikuaatomi asendamine happelise jäägiga. Seetõttu moodustab amiidides olev NH 2 rühm erinevalt amiinidest vaid raskusega ooniumi katiooni. Kuid tugevate hapetega annavad amiidid sooli, näiteks Cl, mida vesi kergesti lagundab. Teisest küljest on amiidides NH 2 rühma vesinik asendatav metallidega kergemini kui ammoniaagis ja amiinides. Näiteks atseetamiid lahustab kergesti elavhõbeoksiidi, moodustades ühendi (CH 3 CONH) 2 Hg.
Siiski on võimalik, et metalliderivaatide moodustumisel toimub amiidi isomerisatsioon ja saadud ühend on imidohappe soola isomeerse (tautomeerse) struktuuriga.
st on olemas analoogia vesiniktsüaniidhappe sooladega.
2. Lämmastikhappe toime Amiidid reageerivad lämmastikhappega, nagu ka primaarsed amiinid, moodustades karboksüülhappeid ja vabastades lämmastikku:
3. Seebistamine Mineraalhapete ja leelistega keetmisel lisavad amiidid vett, moodustades karboksüülhappe ja ammoniaagi:
4. Alküülhalogeniidide toime. Alküülhalogeniidide toime amiididele või nende metalliderivaatidele tekitab N-asendatud amiide:
5. Fosforpentakloriidi toime. Fosforpentakloriidi toimel amiididele tekib klooramiidid
kergesti lagunev vesinikkloriidhappeks ja imiidkloriidid
Viimased koos ammoniaagiga võivad toota sooli amidiinid;
6. Muundamine amiinideks. Amiidide jõulise redutseerimisega võib saada sama arvu süsinikuaatomitega primaarseid amiine:
7. Hoffmanni reaktsioon. Kui amiidid puutuvad kokku hüpohalogeniidi või broomi ja leelisega, tekivad amiinid ja karbonüülrühma süsinikuaatom eraldatakse CO 2 kujul (A. Hoffman). Reaktsiooni kulgu võib kujutada järgmiselt:
Haridusjuhendites leitakse sageli selle reaktsiooni mehhanismi teine tõlgendus:
Kuid see reaktsiooni kulg on fragmendi moodustumise tõttu vähem usutav
kahe vaba elektronpaari kandva lämmastikuaatomiga on ebatõenäoline.
Selle mehhanismi vastu räägib eelkõige asjaolu, et kui radikaal R on optiliselt aktiivne, siis ta reaktsiooni tulemusena ei ratseemiseeru. Vahepeal tooks isegi vaba radikaali R – : üürike olemasolu kaasa optilise aktiivsuse kadumise.
Keemilised omadused. Nitrorühm on üks enim tugevad elektrone väljatõmbavad rühmad ja suudab negatiivset tõhusalt ümber paigutada. tasu. Aromaatsetes ühendus induktiivse ja eriti mesomeerse efekti tulemusena mõjutab see elektrontiheduse jaotust: tuum muutub osaliselt positiivseks. laeng, mis on lokaliseeritud Ch. arr. orto- ja paraasendis; NO 2 rühma Hammetti konstandid s m 0,71, s n 0,778, s + n 0,740, s - n 1,25. Seega suurendab NO 2 rühma sisseviimine reaktsiooni järsult. võime org. ühendus seoses nukleofiilsete reagentidega ja raskendab elektrofiga toimetulekut. reaktiivid. See määrab nitroühendite laialdase kasutamise org. süntees: NO 2 rühm viiakse org-molekuli soovitud asendisse. ühendage, viige läbi lagunemine. funktsioonid, mis on reeglina seotud süsiniku skeleti muutusega ja muudetakse seejärel teiseks funktsiooniks või eemaldatakse. Aromaatsetes Mõnel juhul kasutatakse sageli lühemat skeemi: NO 2 rühma nitreerimine-transformatsioon.
Nitroniühendite moodustumine aromaatsete nitroühendite seerias on seotud benseenitsükli isomeriseerumisega kinoidivormiks; näiteks nitrobenseen moodustub konts. H 2 SO 4 värvilise soolataolise I tüüpi produkt, o-nitrotolueen ilmutab fotokroomsust intramooli tõttu. prootonite ülekanne helesinise O-derivaadi moodustamiseks:
Kui alused toimivad primaarsetele ja sekundaarsetele nitroühenditele, tekivad nitroühendite soolad; soolade ambident-anioonid elektrofiilidega lahustes on võimelised tootma nii O- kui ka C-derivaate. Seega, kui nitroühendite soolad alküülitakse alküülhalogeniidide, trialküülklorosilaanide või R3O + BF-4-ga, tekivad O-alküülimisproduktid. Viimased m.b. saadud ka diasometaani või N,O-bis-(trimetüülsilüül)atseetamiidi toimel nitroalkaanidele pKa-ga< 3 или нитроновые к-ты, напр.:
Atsükliline nitroonhapete alküülestrid on termiliselt ebastabiilsed ja lagunevad moolisiseselt. mehhanism:
R-ts ja s r a r s umbes m-is koos ühendustega ja S-N. Primaarsed ja sekundaarsed nitroühendid kuumutamisel. mineraaliga K-tami on kohal. alkohol või leelise vesilahus moodustavad karbonüülühendeid. (vt Nave reaktsioon). R-tion läbib lünki. nitroniühendite moodustumine:
Algse ühendusena Võib kasutada silüülnitrooneetreid. Tugevate ühendite mõju alifaatsetele nitroühenditele võib põhjustada hüdroksaamühendeid, näiteks:
Nitroühendite redutseerimiseks amiinideks on teada palju meetodeid. Laialdaselt kasutatakse rauaviile, Sn ja Zn. komplekt; katalüütikuga hüdrogeenimisel kasutatakse katalüsaatoritena Ni-Raney, Pd/C või Pd/PbCO 3 jt. Alifaatsed nitroühendid redutseeritakse juuresolekul kergesti amiinideks LiAlH 4 ja NaBH 4. Pd, Na ja Al amalgaamid kuumutamisel. hüdrasiiniga Pd/C kohal; aromaatsete nitroühendite puhul kasutatakse mõnikord TlCl 3, CrCl 2 ja SnCl 2, aromaatsed. polünitroühendid redutseeritakse selektiivselt nitramiinideks Na hüdrosulfiidi toimel CH 3 OH-s. Valimiseks on viise. NO 2 rühma redutseerimine polüfunktsionaalsetes nitroühendites teisi funktsioone mõjutamata.
Kui P(III) mõjutab aromaatseid nitroühendeid, tekib järjestus. NO 2 rühma deoksüdeerimine koos väga reaktiivsete nitreenide moodustumisega. Lahust kasutatakse kondensaatori sünteesiks. heterotsüklid, näiteks:
R-ts ja NO 2 rühma säilimine. A-H aatomit sisaldavad alifaatsed nitroühendid on kergesti alküülitud ja atsüülitavad, moodustades tavaliselt O-derivaate. Küll aga vastastikune mod. primaarsete nitroühendite dilitiumisoolad alküülhalogeniidide, anhüdriidide või karboksüülhapete halogeniididega põhjustavad C-alküülimis- või C-atsüülimisprodukte, näiteks:
Intramooli näiteid on teada. C-alküülimine, nt:
Primaarsed ja sekundaarsed nitroühendid reageerivad alifaatsete ühenditega. amiinid ja CH2O koos p-aminoderivaatide moodustumisega (Mannichi lahus); lahuses võite kasutada eelnevalt valmistatud nitroühendite või aminoühendite metülooli derivaate:
Nitrometaan ja nitroetaan võivad kondenseeruda kahe metüloolamiini molekuliga ja kõrgemad nitroalkaanid ainult ühe molekuliga. Teatud reagentide vahekorras võib lahus viia heterotsükliliseks. ühendus, näiteks: suhtlemisel moodustub primaarne nitroalkaan koos kahe ekvivalendi primaarse amiini ja liigse formaldehüüdiga. Vormid V, kui reaktiive võetakse vahekorras 1:1:3-comm. Vormid VI.
Aromaatsed nitroühendid sisenevad kergesti nukleofiilsetesse lahustesse. asendamine ja palju raskem - elektrofi piirkonnas. asendamine; sel juhul suunatakse nukleofiil orto- ja po-positsioonidesse ning elektrofiil on suunatud NO 2 rühma meta-asendisse. Elektroonilise kiiruse konstant nitrobenseeni nitreerimine on 5-7 suurusjärku väiksem kui benseeni oma; see tekitab m-dinitrobenseeni.
Primaarsete nitroalkaanide karboksüülimisel CH 3 OMgOCOOCH 3 toimel tekivad a-lämmastikuühendid või nende estrid.
Kui mononitroühendite C(NO 2) 4 sooli töödeldakse Ag või leelismetalli nitrititega või kui nitritid mõjutavad leeliselises keskkonnas (Ter Meeri lahus) a-halo-nitroalkaane, tekivad heem-dinitroühendid. a-halogeno-nitroalkaanid aprotoonsetes lahustes, samuti nitroühendite Cl2 töötlemine leeliselises keskkonnas või nitroühendite soolade elektrooksüdeerimine põhjustavad vic-dinitroühendeid:
Nitrorühm ei renderda olendeid. mõju aromaatsete ühendite vabade radikaalide alküülimisele või arüülimisele. ühendus; r-tion viib alusele. orto- ja paraasendatud toodetele.
Nitroühendite vähendamiseks ilma NO 2 rühma mõjutamata kasutage NaBH 4, LiAlH 4 madalatel temperatuuridel või diboraani lahust THF-s, näiteks:
Aromaatne di- ja trinitroühendid, eriti 1,3,5-trinitrobenseen, moodustavad stabiilseid erksavärvilisi kristalseid ühendeid. nad ütlesid kompleksid aromaatsusega elektronidoonorühendid (amiinid, fenoolid jne). Aromaatsete ühendite eraldamiseks ja puhastamiseks kasutatakse komplekse pikriinhappega. süsivesinikud. Interaktsioon Tugevate alustega di- ja trinitrobenseenid (HO - , RO - , N - 3 , RSO - 2 , CN - , alifaatsed amiinid) põhjustavad Meisen-Haimeri komplekside moodustumist, mis eraldatakse värviliste leelismetallisoolade kujul.
Nende reaktsioonide jaoks sobivad oksüdeerivad ained on kroom- või lämmastikhape, kroomisegu, mangaandioksiid või seleenidioksiid.
Kroomihappega oksüdeerimisel kinnitub alkohol nukleofiilselt kroomhappega, mille käigus eraldub vesi ja moodustub kroomhappe ester (see on reaktsiooni esimene etapp, sarnaneb karboksüülhapete estrite moodustumisega). vt punkt E, 7.1.5.1). Teises etapis, mis tõenäoliselt läbib tsüklilise ülemineku oleku, läheb alkoholi a-vesinik kromaadijäägiks ja metall kuuevalentsest olekust neljavalentsesse:
| n-CH30> P-tert-C4H9> P-CH3> P-Cl> P- EI 2 | (G.6.20) |
Primaarsete alkoholide oksüdeerimisel tuleb tekkivat aldehüüdi kaitsta edasise oksüdeerumise eest karboksüülhappeks. Näiteks on võimalik reaktsioonisegust aldehüüdi pidevalt välja destilleerida: see on täiesti teostatav, kuna aldehüüdi keemistemperatuur on tavaliselt madalam kui vastava alkoholi keemistemperatuur. Sellegipoolest ületab aldehüüdide saagis dikromaadiga oksüdeerimisel harva 60%. Tähelepanuväärne on, et kui reaktsioon on korralikult läbi viidud, ei mõjuta mitmed süsinik-süsinik sidemed peaaegu.
Aldehüüdid tekivad ka alkoholide kuumutamisel neutraalse dikromaadi vesilahusega, kuid ainult bensüülalkoholid annavad hea saagise.
Aldehüüdide suuremat saagist saab primaarsete alkoholide oksüdeerimisel hõõrub-butüülkromaat (petrooleetris, benseenis või süsiniktetrakloriidis) või mangaandioksiid (atsetoonis, petrooleetris, süsiniktetrakloriidis või lahjendatud väävelhappes). Need reaktiivid võimaldavad saada hea saagisega ka küllastumata ja aromaatseid aldehüüde.
Sekundaarsete alkoholide oksüdeerimine ketoonideks on isegi lihtsam kui primaarsete alkoholide oksüdeerimine. Siin on saagised suuremad, sest esiteks on sekundaarsete alkoholide reaktsioonivõime kõrgem kui primaarsetel alkoholidel ja teiseks on tekkivad ketoonid aldehüüdidega võrreldes palju vastupidavamad oksüdatsioonile. Steroidide ja terpeenide hulgas on ennast tõestanud sekundaarsete alkoholide oksüdeerimine kroomhappe ja püridiini kompleksiga, samuti kroomanhüdriidiga dimetüülformamiidis. Atsetoonis olev kroomanhüdriid on samuti hea oksüdeerija; seda saab kasutada küllastumata sekundaarsete alkoholide oksüdeerimiseks, ilma et see mõjutaks mitmekordset süsinik-süsinik sidet.
Uus meetod, mis sobib ka steeriliselt takistatud alkoholide jaoks, on oksüdeerimine dimetüülsulfoksiidiga äädikhappe anhüdriidis.
Vastavalt allpool toodud protseduurile viiakse reaktsioon läbi kahefaasilises süsteemis. Saadud ketoonid ekstraheeritakse orgaanilise lahustiga ja on seega kaitstud edasise oksüdatsiooni eest.
Disahhariidid– süsivesikud, mille molekulid koosnevad kahest monosahhariidijäägist, mis on omavahel seotud kahe hüdroksüülrühma interaktsiooni kaudu.
Disahhariidimolekuli moodustumise ajal elimineeritakse üks veemolekul:
või sahharoosi jaoks:
Seetõttu on disahhariidide molekulaarne valem C 12 H 22 O 11.
Sahharoosi moodustumine toimub taimerakkudes ensüümide mõjul. Kuid keemikud on leidnud viisi, kuidas viia läbi paljusid reaktsioone, mis on osa eluslooduses toimuvatest protsessidest. 1953. aastal viis prantsuse keemik R. Lemieux esmakordselt läbi sahharoosi sünteesi, mida kaasaegsed nimetasid "orgaanilise keemia Everesti vallutamiseks".
Tööstuses saadakse sahharoosi suhkruroo mahlast (sisaldus 14-16%), suhkrupeedist (16-21%), aga ka mõnest teisest taimest, näiteks kanada vahtrast või maapirnist.
Kõik teavad, et sahharoos on kristalne aine, millel on magus maitse ja mis lahustub vees hästi.
Suhkruroomahl sisaldab süsivesikuid sahharoosi, mida tavaliselt nimetatakse suhkruks.
Saksa keemiku ja metallurgi A. Marggrafi nimi on tihedalt seotud peedist suhkru valmistamisega. Ta oli üks esimesi teadlasi, kes kasutas oma keemiauuringutes mikroskoopi, millega avastas 1747. aastal peedimahlas suhkrukristallid.
Laktoos - kristalne piimasuhkur, saadi 17. sajandil imetajate piimast. Laktoos on vähem magus disahhariid kui sahharoos.
Nüüd tutvume süsivesikutega, millel on keerulisem struktuur - polüsahhariidid.
Polüsahhariidid– suure molekulmassiga süsivesikuid, mille molekulid koosnevad paljudest monosahhariididest.
Lihtsustatud kujul võib üldise skeemi esitada järgmiselt:
Nüüd võrdleme tärklise ja tselluloosi - polüsahhariidide olulisemate esindajate - struktuuri ja omadusi.
Nende polüsahhariidide polümeeriahelate struktuuriüksuseks, mille valem on (C 6 H 10 O 5) n, on glükoosijäägid. Struktuuriüksuse koostise (C 6 H 10 O 5) kirja panemiseks tuleb glükoosi valemist lahutada veemolekul.
Tselluloos ja tärklis on taimset päritolu. Need moodustuvad polükondensatsiooni tulemusena glükoosi molekulidest.
Polükondensatsioonireaktsiooni võrrandi ja selle polüsahhariidide pöördhüdrolüüsi protsessi saab tinglikult kirjutada järgmiselt:
Tärklise molekulidel võib olla nii lineaarne kui ka hargnenud struktuur, tselluloosi molekulidel võib olla ainult lineaarne struktuur.
Joodiga suhtlemisel annab tärklis erinevalt tselluloosist sinise värvi.
Neil polüsahhariididel on ka taimerakkudes erinevad funktsioonid. Tärklis toimib varutoitainena, tselluloos täidab struktuurset, ehituslikku funktsiooni. Taimerakkude seinad on valmistatud tselluloosist.
KANSITSAROREAKTSIOON, oksüdatsioon-vähendamine aldehüüdide disproportsioon leelise mõjul primaarsete alkoholide ja süsihapete moodustumisega, näiteks:
Aldehüüdi töödeldakse konts. leelise vesi- või vesi-alkohollahus jahutamisel või kergelt kuumutamisel Katalüsaatorid - lagunevad. metallid (nt Ag, Ni, Co, Cu) ja nende oksiidid. Lahusesse sisenevad aldehüüdid, mis ei sisalda karbonüülrühma a-asendis H-aatomit. Vastasel juhul ei ole eelistatav mitte Cannizzaro reaktsioon, vaid pigem aldooli kondensatsioon. Aromaatse ringi elektrone eemaldavad asendajad. aldehüüdid kiirendavad protsessi ja elektrone loovutavad aeglustavad. Bensaldehüüdid, mille asendajad on orto-asendis, ei reageeri Cannizzaros; o- ja p-hüdroksübensaldehüüdid reageerivad ainult nende juuresolekul. Ag. Peatükis kasutatakse reaktsiooni, milles kasutatakse kahte erinevat aldehüüdi (nn rist-Cannizzaro reaktsioon). arr. primaarsete alkoholide saamiseks aromaatsetest ainetest suure saagisega. aldehüüdid. Redutseerijana kasutatakse tavaliselt formaldehüüdi:
ArCHO + CH 2 O: ArCH 2 OH + HCOOH
Polühüdroksümetüleeritud ühendite sünteesi käigus. formaldehüüd osaleb esimesel etapil aldooli kondenseerumisel ja seejärel redutseerijana Cannizzaro ristreaktsioonis:
Cannizzaro pakutud homogeense reaktsiooni mehhanism. keskkond sisaldab hüdriidi ülekandeetappi
Aromaatseks aldehüüdid, ei saa välistada ühe elektroni ülekande tulemusena tekkinud radikaalanioonide Cannizzaro reaktsioonis osalemise võimalust. Intramooli kasutamisel tekib Cannizzaro reaktsiooniga sarnane reaktsioon. a-ketoaldehüüdide disproportsioon. leelised (Cannizzaro ümberkorraldus):
Cannizzaro reaktsiooni kasutatakse tööstuslikel eesmärkidel. pentaerütritooli süntees, alkoholide, süsinikuühendite jne ettevalmistav tootmine Protsessi avastas S. Cannizzaro 1853. aastal.
Pürrool, furaan ja tiofeen on ühe heteroaatomiga viieliikmelised heterotsüklilised ühendid.
Aatomite nummerdamine heterotsüklis algab heteroaatomist ja kulgeb vastupäeva. Asendeid 2 ja 5 nimetatakse a-positsioonideks, 3 ja 4 b-positsioonideks.
Formaalsete omaduste järgi klassifitseeritakse need ühendid aromaatseteks, kuna need on konjugeeritud tsüklilised p-süsteemid, mis sisaldavad 6p elektroni - 4 dieenisüsteemi elektroni - ja heteroaatomi elektronide paari. Tsükkel on peaaegu tasane, mis tähendab, et heteroaatomi hübridisatsiooni olek on lähedane sp 2-le.
Allpool on toodud resonantsstruktuurid, mis illustreerivad heteroaatomi elektronide ümberpaigutamist piki heterotsüklilist ringi, kasutades näitena furaani.
Ülaltoodud resonantsstruktuurid näitavad, et heteroaatom (antud juhul hapnikuaatom) kannab dieeni π-süsteemiga mesomeerse interaktsiooni tulemusena tsüklile üle elektrontiheduse, mille tulemusena tekib tsüklile teatav negatiivne laeng. süsinikuaatomid heterotsüklis ja vastavalt positiivne laeng hapnikuaatomi laengule. Hapnikuaatomil on lisaks positiivsele mesomeersele efektile loomulikult ka negatiivne induktiivne toime. Selle avaldumine vaadeldavate ühendite omadustes on aga vähem väljendunud ja seetõttu liigitatakse ühe heteroaatomiga viieliikmelised heterotsüklid p-liigsete aromaatsete heterotsükliliste ühendite hulka. Resonants viib heterotsüklis sidemepikkuste mõningase ühtlustumiseni, mis viitab ka süsteemi teatud aromaatsusele.
MÄÄRATLUS
Orgaanilised ühendid, mis on karboksüülhapete derivaadid, mis tekivad viimaste interaktsioonil alkoholidega:
Estrite üldine struktuurivalem:

kus R ja R’ on süsivesinikradikaalid.
Estrite hüdrolüüs
Estrite üks iseloomulikumaid omadusi (lisaks esterdamisele) on nende hüdrolüüs – lõhustumine vee mõjul. Teisel viisil nimetatakse estrite hüdrolüüsi seebistamiseks. Erinevalt soolade hüdrolüüsist on see antud juhul praktiliselt pöördumatu. Eristatakse estrite aluselist ja happelist hüdrolüüsi. Mõlemal juhul moodustuvad alkohol ja hape:
a) happeline hüdrolüüs

b) leeliseline hüdrolüüs

Näited probleemide lahendamisest
NÄIDE 1
| Harjutus | Määrake äädikhappe mass, mida on võimalik saada 180 g kaaluva etüülatsetaadi seebistamisreaktsiooni käigus. |
| Lahendus | Kirjutame äädikhappe etüülestri hüdrolüüsi reaktsioonivõrrandi brutovalemiga: C 4 H 8 O 2 + H 2 O ↔ CH 3 COOH + C 2 H 5 OH. Arvutame etüülatsetaadi aine koguse (moolmass - 88 g/mol), kasutades probleemitingimustest saadud massi väärtust: υ (C4H8O2) = m (C4H8O2)/M (C4H8O2) = 180/88 = 2 mol. Reaktsioonivõrrandi järgi on etüülatsetaadi ja äädikhappe moolide arv võrdne: υ (C4H8O2) = υ (CH3COOH) = 2 mol. Seejärel saate määrata äädikhappe massi (moolmass - 60 g/mol): m(CH3COOH) = υ (CH3COOH) × M (CH3COOH) = 2 × 60 = 120 g. |
| Vastus | Äädikhappe mass on 120 grammi. |
Estrite hüdrolüüsi katalüüsivad nii happed kui ka alused. Estrite happeline hüdrolüüs viiakse tavaliselt läbi kuumutamisel vesinikkloriid- või väävelhappega vesi- või vesi-alkoholikeskkonnas. Orgaanilises sünteesis kasutatakse estrite happelist hüdrolüüsi kõige sagedamini mono- ja dialküülasendatud maloonestrite puhul (17. peatükk). Maloonestri mono- ja diasendatud derivaadid läbivad kontsentreeritud vesinikkloriidhappega keetmisel hüdrolüüsi, millele järgneb dekarboksüülimine.
Alusega katalüüsitud hüdrolüüsiks kasutatakse tavaliselt NaOH või KOH vesi- või vesi-alkoholilahust. Parimad tulemused saavutatakse kaaliumhüdroksiidi õhukese suspensiooni kasutamisel DMSO-s, mis sisaldab väikest kogust vett.

Viimast meetodit eelistatakse takistatud happe estrite seebistamiseks; selle meetodi teiseks modifikatsiooniks on takistatud estrite aluseline hüdrolüüs 18-kroon-6-polüestri juuresolekul:

Preparatiivsetel eesmärkidel on aluskatalüüsitud hüdrolüüsil happelise hüdrolüüsi ees mitmeid ilmseid eeliseid. Estrite aluselise hüdrolüüsi kiirus on tavaliselt tuhat korda suurem kui happekatalüüsil. Hüdrolüüs happelises keskkonnas on pöörduv protsess, erinevalt hüdrolüüsist aluse juuresolekul, mis on pöördumatu.
18.8.2.A. Estri hüdrolüüsi mehhanismid
Estrite hüdrolüüs puhta veega on enamikul juhtudel pöörduv reaktsioon, mille tulemuseks on karboksüülhappe ja lähteestri tasakaaluline segu:

See reaktsioon kiireneb oluliselt happelises ja leeliselises keskkonnas, mis on seotud happe-aluse katalüüsiga (3. peatükk).
K. Ingoldi järgi klassifitseeritakse estri hüdrolüüsi mehhanismid järgmiste kriteeriumide järgi:
(1) Katalüüsi tüüp: happeline (tähis A) või aluseline (tähis B);
(2) Lõhustamise tüüp, mis näitab, milline kahest estri C-O -sidemest reaktsiooni tulemusena lõhustub: atsüülhapnik (AC-indeks) või alküülhapnik (AL-indeks):

(3) Reaktsiooni molekulaarsus (1 või 2).
Nendest kolmest kriteeriumist saab teha kaheksa erinevat kombinatsiooni, mis on näidatud diagrammil 18.1.

Need on kõige levinumad mehhanismid. Leeliseline seebistamine kuulub peaaegu alati tüüpi B AC 2. Happelisel hüdrolüüsil (nagu ka esterdamisel) on enamikul juhtudel mehhanism A AC 2.

Mehhanism A AC1 täheldatakse tavaliselt ainult tugevalt happelistes lahustes (näiteks kontsentreeritud H2SO4-s) ja eriti sageli steeriliselt takistatud aromaatsete happe estrite puhul.
AC 1 mehhanism on siiani teadmata.

BAL 2 mehhanism leiti ainult erakordselt tugevate ruumiliselt varjestatud atsüülrühmade ja β-laktoonide neutraalse hüdrolüüsi korral. A AL 2 mehhanism on siiani teadmata.

Vastavalt A AL 1 mehhanismile reageerivad tertsiaarsed alküülestrid tavaliselt neutraalses või happelises keskkonnas. Samad substraadid sarnastes tingimustes võivad reageerida vastavalt B AL 1 mehhanismile, kuid veidi aluselisemasse keskkonda liikudes asendub B AL 1 mehhanism kohe B AC 2 mehhanismiga.
Nagu skeemilt 18.1 näha, on hapete poolt katalüüsitud reaktsioonid pöörduvad ning mikroskoopilise pöörduvuse põhimõttest (peatükk 2) järeldub, et hapete poolt katalüüsitav esterdamine toimub samuti sarnaste mehhanismide kaudu. Aluskatalüüsiga aga nihkub tasakaal hüdrolüüsi (seebistamise) suunas, kuna tasakaal nihkub karboksüülhappe ionisatsiooni tõttu. Vastavalt ülaltoodud skeemile on mehhanismi A korral AC1 rühmad COOR ja COOH protoneeritud alkoksü- või hüdroksüülhapniku aatomi juures. Üldiselt on termodünaamika seisukohalt soodsam karbonüülhapniku, C=O rühma protoneerimine, kuna sel juhul saab positiivse laengu kahe hapnikuaatomi vahel ümber paigutada:

Sellele vaatamata sisaldab lahus väikestes kogustes ka tautomeerset katiooni – vajalikku vaheühendit A AC 1 mehhanismis.Mõlemad B1 mehhanismid (millest B AC 1 on teadmata) ei ole tegelikult üldse katalüütilised, sest alguses dissotsieerub tekib neutraalne ester.
Kaheksast Ingoldi mehhanismist on eksperimentaalselt tõestatud vaid kuus.