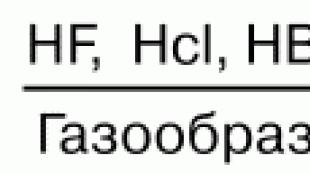Jedným z produktov hydrolýzy esterov je. Hydrolýza esterov
Estery sú funkčné deriváty karboxylových kyselín so všeobecným vzorcom RC(0)OR."
Spôsoby získavania. Najvýznamnejším spôsobom získania esterov je acylácia alkoholov a fenolov rôznymi acylačnými činidlami, napríklad karboxylovými kyselinami, chloridmi kyselín, anhydridmi. Možno ich získať aj Tiščenkovou reakciou.
Estery sa pripravujú vo vysokých výťažkoch alkyláciou solí karboxylových kyselín alkylhalogenidmi:
Estery vznikajú elektrofilnou adíciou karboxylových kyselín na alkény a alkíny. Reakcia sa často používa na výrobu esterov terciárnych alkoholov, napr. drhne-butylétery:
Pridaním kyseliny octovej k acetylénu vzniká priemyselne dôležitý monomér vinylacetát, Octan zinočnatý na aktívnom uhlí sa používa ako katalyzátor:
Hydrolýza. Najdôležitejšou z acylačných reakcií je hydrolýza esterov za vzniku alkoholu a karboxylovej kyseliny:
Reakcia prebieha v kyslom aj alkalickom prostredí. Kyselina katalyzovaná hydrolýza esterov - reverzná reakcia esterifikácie prebieha rovnakým mechanizmom Als 2
Alkalická hydrolýza je nevratná; počas reakcie sa spotrebuje mól alkálie na mól éteru, t.j. alkália v tejto reakcii pôsobí ako spotrebné činidlo a nie ako katalyzátor:
Hydrolýza esterov v alkalickom prostredí prebieha bimolekulárnym acylovým mechanizmom VAS2 cez štádium tvorby tetraedrického intermediátu (I). Ireverzibilnosť alkalickej hydrolýzy je zabezpečená prakticky ireverzibilnou acidobázickou interakciou karboxylovej kyseliny (I) a alkoxidového iónu (III). Výsledný anión karboxylovej kyseliny (IV) je sám o sebe dosť silný nukleofil, a preto nepodlieha nukleofilnému ataku.
Transesterifikácia. Pomocou tejto reakcie sa vzájomná konverzia esterov tej istej kyseliny uskutočňuje podľa nasledujúcej schémy:
Transesterifikácia je reverzibilný proces, katalyzovaný kyselinami aj zásadami a prebieha rovnakým mechanizmom ako reakcie esterifikácie a hydrolýzy esterov. Rovnováha sa posúva dobre známymi metódami, a to použitím prebytku reakčného alkoholu (R"OH vo vyššie uvedenom diagrame - pre posun doprava) alebo oddestilovaním jedného z reakčných produktov, ak má najnižšiu teplotu varu. Transesterifikáciou sa získa napríklad dobre známe anestetikum novokaín(báza) z etylesteru kyseliny l-aminobenzoovej:
Esterová kondenzácia. Keď dve molekuly esteru kondenzujú v prítomnosti zásaditého katalyzátora, vytvoria sa estery β-oxokyselín:
Molekula etylacetátu má slabé vlastnosti CH-kyseliny v dôsledku indukčného účinku esterovej skupiny a je schopná interagovať so silnou zásadou - etoxidovým iónom:
Amidy karboxylových kyselín. Spôsoby získavania. Štruktúra amidovej skupiny. Acidobázické vlastnosti amidov. Kyslá a alkalická hydrolýza. Štiepenie amidov halogénmi v alkalickom prostredí a kyselinou dusitou. Dehydratácia na nitrily.
Amidy sú funkčné deriváty karboxylových kyselín všeobecného vzorca R-C(O)-NH2_nR", kde P = 0-2.
Spôsoby získavania. Najdôležitejším spôsobom prípravy amidov je acylácia amoniaku a amínov halogenidmi, anhydridmi a estermi kyselín.
Acylácia amoniaku a amínov halogenidmi kyselín. Acylačná reakcia amoniaku a amínov s halogenidmi kyselín je exotermická a uskutočňuje sa po ochladení:
Acylácia amoniaku a amínov anhydridmi. Na acetyláciu amínov sa najčastejšie používa najdostupnejší anhydrid, acetanhydrid:
Amonolýza esterov. Aminy sa získavajú amonolýzou esterov. Napríklad, keď vodný roztok amoniaku reaguje s dietylfumarátom, vytvorí sa úplný amid kyseliny fumarovej:
Štruktúra amidov. Elektrónová štruktúra amidovej skupiny je do značnej miery podobná štruktúre karboxylovej skupiny. Amidová skupina je p,l-konjugovaný systém, v ktorom je osamotený pár elektrónov atómu dusíka konjugovaný s elektrónmi väzby C=0. Delokalizácia elektrónovej hustoty v amidovej skupine môže byť reprezentovaná dvoma rezonančnými štruktúrami:
V dôsledku konjugácie je väzba C-N v amidoch čiastočne dvojito viazaná; jej dĺžka je výrazne menšia ako dĺžka jednoduchej väzby v amínoch, zatiaľ čo väzba C=0 je o niečo dlhšia ako väzba C=0 v aldehydoch a ketónoch. Amidová skupina má plochú konfiguráciu v dôsledku konjugácie. Nižšie sú uvedené geometrické parametre molekuly iV-substituovaného amidu, stanovené pomocou rôntgenovej difrakčnej analýzy:
Acidobázické vlastnosti. Amidy majú slabé kyslé a zásadité vlastnosti. Zásaditosť amidov leží v rozmedzí hodnôt pA"in+ od -0,3 do -3,5. Dôvodom zníženej zásaditosti aminoskupiny v amidoch je konjugácia osamelého páru elektrónov atómu dusíka s karbonylom Pri interakcii so silnými kyselinami sa amidy protónujú na atóme kyslíka ako v zriedených a koncentrovaných roztokoch kyselín. Tento druh interakcie je základom kyslej katalýzy pri hydrolytických reakciách amidov:
Acylačné reakcie. V dôsledku prítomnosti silnej elektrón-donornej aminoskupiny v konjugovanom amidovom systéme je elektrofilita karbonylového uhlíkového atómu, a teda reaktivita amidov pri acylačných reakciách, veľmi nízka. Nízka acylačná schopnosť amidov sa vysvetľuje aj skutočnosťou, že amidový ión NH2- je slabo odstupujúcou skupinou. Spomedzi acylačných reakcií má praktický význam hydrolýza amidov, ktorá sa môže uskutočňovať v kyslom a alkalickom prostredí. Amidy sa hydrolyzujú oveľa ťažšie ako iné funkčné deriváty karboxylových kyselín. Hydrolýza amidov sa uskutočňuje za prísnejších podmienok v porovnaní s hydrolýzou esterov.
Kyslá hydrolýza amidov je ireverzibilná reakcia vedúca k tvorbe karboxylovej kyseliny a amónnej soli:
Alkalická hydrolýza je tiež nevratná reakcia; v dôsledku toho sa vytvorí soľ karboxylovej kyseliny a amoniak alebo amín:
Trávenie kyseliny dusičnej. Pri interakcii s kyselinou dusitou a inými nitrozačnými činidlami sa amidy premieňajú na zodpovedajúce karboxylové kyseliny s výťažkom až 90 %:
Kyselina uhličitá a jej funkčné deriváty; fosgén, chlóruhľovodíkové étery, kyselina karbámová a jej estery (uretány). Močovina (močovina), základné a nukleofilné vlastnosti. Hydrolýza močoviny. Acylmočoviny (ureidy), kyseliny ureidové. Interakcia močoviny s kyselinou dusitou a brómanmi. Guanidín, základné vlastnosti.
Kyselina uhličitá tradične nepatrí medzi organické zlúčeniny, ale ona sama a jej funkčné deriváty majú určitú podobnosť s karboxylovými kyselinami a ich derivátmi, a preto sú diskutované v tejto kapitole.
Kyselina dvojsýtna uhličitá je nestabilná zlúčenina, ktorá sa ľahko rozkladá na oxid uhličitý a vodu. Vo vodnom roztoku oxidu uhličitého existuje iba 0,1 % vo forme kyseliny uhličitej. Kyselina uhličitá tvorí dve série funkčných derivátov – úplné (stredné) a neúplné (kyslé). Estery kyselín, amidy a iné deriváty sú nestabilné a rozkladajú sa za vzniku oxidu uhličitého:
Kompletný chlorid uhličitý - fosgén COC1 2 - je nízkovriaca kvapalina so zápachom zhnitého sena, veľmi toxická, spôsobuje pľúcny edém a vzniká ako škodlivá nečistota pri fotochemickej oxidácii chloroformu v dôsledku nesprávneho skladovania. posledne menované.
V priemysle sa fosgén vyrába radikálovou chloráciou oxidu uhoľnatého (II) v reaktore naplnenom aktívnym uhlím:
Fosgén, podobne ako kyslé chloridy karboxylových kyselín, má vysokú acylačnú schopnosť, získavajú sa z neho mnohé ďalšie funkčné deriváty kyseliny uhličitej.
Keď fosgén reaguje s alkoholmi, vytvárajú sa dva typy esterov - úplné (uhličitany) a čiastočné (estery chloridu uhličitého alebo chlórformiáty), pričom posledné sú estery aj chloridy kyselín. Ako akceptor chlorovodíka a nukleofilný katalyzátor sa používajú terciárne amíny alebo pyridín.
Kyselina karbamová- neúplný amid kyseliny uhličitej - nestabilná zlúčenina, rozkladá sa za vzniku amoniaku a oxidu uhličitého:
Estery kyseliny karbamovej - karbamáty, alebo uretány, - stabilné zlúčeniny získané pridaním alkoholov k izokyanátom alebo acyláciou amoniaku a amínov zodpovedajúcim chlórformiátom:
Močovina(karbamid) - úplný amid kyseliny uhličitej - prvýkrát izoloval z moču I. Ruel (1773). Je najdôležitejším konečným produktom metabolizmu bielkovín u cicavcov; dospelý človek vylúči 25-30 g močoviny denne. Močovinu prvýkrát syntetizoval F. Wöhler (1828) zahrievaním kyanátu amónneho:
Táto syntéza bola prvým príkladom získania organickej látky z anorganickej zlúčeniny.
V priemysle sa močovina vyrába z amoniaku a oxidu uhličitého pri zvýšenom tlaku a teplote (180 – 230 °C, 150 – 200 atm):
Močovina má slabé zásadité vlastnosti (p.iHvn + 0,1) a so silnými kyselinami tvorí soli. Soli kyseliny dusičnej a šťaveľovej sú nerozpustné vo vode.
Močovina je protónovaná skôr na atóme kyslíka ako na atóme dusíka. Je to pravdepodobne spôsobené delokalizáciou osamelých párov elektrónov atómov dusíka v dôsledku konjugácie p,π.
Vo vriacej vode sa močovina hydrolyzuje za vzniku amoniaku a oxidu uhličitého; kyseliny a zásady katalyzujú túto reakciu:
Primárne produkty vznikajúce pri zahrievaní močoviny sú amoniak a kyselina izokyanát. Kyselina izokyaná môže trimerizovať na kyselinu kyanurovú alebo kondenzovať s druhou molekulou močoviny za vzniku biuretu. V závislosti od rýchlosti ohrevu dominuje jedna alebo druhá cesta rozkladu močoviny:
Pôsobením hypohalitov dochádza aj k rozkladu močoviny. V závislosti od podmienok sa môže tvoriť dusík alebo hydrazín; To je presne to, ako sa ten druhý získava v priemysle:
Močovina tiež vykazuje nukleofilné vlastnosti pri alkylačných a acylačných reakciách. Alkylácia močoviny, v závislosti od alkylačného činidla, môže viesť k O- a TV-alkyl derivátom:
Guanidín alebo iminomočovina (H 2 N) 2 C=NH sa priemyselne vyrába fúziou močoviny s dusičnanom amónnym alebo zahrievaním esterov kyseliny ortokarbónovej s amoniakom:
Guanidín je bezfarebná kryštalická látka so silnými zásaditými vlastnosťami. Vysoká zásaditosť na úrovni hydroxidov alkalických kovov je spôsobená úplnou delokalizáciou kladného náboja v symetrickom guanidíniovom katióne:
Guanidínové a biguanidínové zvyšky sa nachádzajú v niektorých prírodných zlúčeninách a liečivých látkach.
Estery sa nazývajú funkčné deriváty karboxylových kyselín všeobecného vzorca RC(O)OR" .
Estery karboxylových kyselín (rovnako ako sulfónové kyseliny) sa nazývajú podobne ako soli, len namiesto názvu katiónu sa používa názov zodpovedajúceho alkylu alebo arylu, ktorý sa umiestňuje pred názov aniónu a píše sa spolu s to. Prítomnosť esterovej skupiny -COOR sa môže prejaviť aj opisným spôsobom, napríklad „R-ester (takej a takej) kyseliny“ (táto metóda je menej výhodná kvôli jej ťažkopádnosti):
Estery nižších alkoholov a karboxylových kyselín sú prchavé kvapaliny s príjemnou vôňou, zle rozpustné vo vode a dobre rozpustné vo väčšine organických rozpúšťadiel. Vône esterov pripomínajú vône rôznych druhov ovocia, preto sa v potravinárskom priemysle používajú na prípravu esencií, ktoré napodobňujú ovocné vône. Zvýšená prchavosť esterov sa využíva na analytické účely.
Hydrolýza. Najdôležitejšou z acylačných reakcií je hydrolýza esterov za vzniku alkoholu a karboxylovej kyseliny:
Reakcia prebieha v kyslom aj alkalickom prostredí. Kyselina katalyzovaná hydrolýza esterov - reverzná reakcia na esterifikáciu, prebieha podľa rovnakého mechanizmu A AC 2:
Nukleofilom v tejto reakcii je voda. Posun rovnováhy smerom k tvorbe alkoholu a kyseliny je zabezpečený pridaním prebytočnej vody.
Alkalická hydrolýza je nevratná; počas reakcie sa spotrebuje mól alkálie na mól éteru, t.j. alkália v tejto reakcii pôsobí ako spotrebné činidlo a nie ako katalyzátor:
Hydrolýza esterov na alkalické prostredie prebieha bimolekulárnym acylovým mechanizmom B AC 2 cez štádium tvorby tetraedrického intermediátu (I). Ireverzibilnosť alkalickej hydrolýzy je zabezpečená prakticky ireverzibilnou acidobázickou interakciou karboxylovej kyseliny (II) a alkoxidového iónu (III). Výsledný anión karboxylovej kyseliny (IV) je sám o sebe dosť silný nukleofil, a preto nepodlieha nukleofilnému ataku.
Amonolýza esterov. Aminy sa získavajú amonolýzou esterov. Napríklad, keď vodný roztok amoniaku reaguje s dietylfumarátom, vytvorí sa úplný amid kyseliny fumarovej:
Počas amonolýzy esterov s amínmi s nízkou nukleofilitou sa tieto najskôr premenia na amidy alkalických kovov alebo kovov alkalických zemín:

Amidy karboxylových kyselín: nomenklatúra; štruktúra amidovej skupiny; acidobázické vlastnosti; kyslá a alkalická hydrolýza; štiepenie bromňanmi a kyselinou dusitou; dehydratácia na nitrily; chemická identifikácia.
amidy sa nazývajú funkčné deriváty karboxylových kyselín všeobecného vzorca R-C(0)-NH2-nR"n, kde n = 0-2. V nesubstituovaných amidoch je acylový zvyšok pripojený k nesubstituovanej aminoskupine, v N-substituovaných amidoch je jeden z atómov vodíka nahradený jedným alkylovým alebo arylovým zvyškom, v N,N-substituovaných amidoch dvoma.
Zlúčeniny obsahujúce jednu, dve alebo tri acylové skupiny pripojené k atómu dusíka sa genericky nazývajú amidy (primárne, sekundárne a terciárne). Názvy primárnych amidov s nesubstituovanou skupinou - NH 2 sú odvodené od názvov zodpovedajúcich acylových zvyškov nahradením prípony -olej (alebo -yl) -amidom. Amidy tvorené z kyselín s príponou -karboxylová kyselina dostávajú príponu -karboxamid. Amidy kyseliny sulfónovej sú tiež pomenované podľa zodpovedajúcich kyselín pomocou prípony -sulfónamid.
Názvy radikálov RCO-NH- (ako RSO 2 -NH-) sú tvorené z názvov amidov, pričom sa mení prípona -amid na -amido-. Používajú sa, ak zvyšok molekuly obsahuje vyššiu skupinu alebo k substitúcii dochádza v zložitejšej štruktúre ako radikál R:

V názvoch N-substituovaných primárnych amidov RCO-NHR" a RCO-NR"R" (ako aj podobných sulfónamidov) sú názvy zvyškov R" a R" uvedené pred názvom amidu so symbolom N -:
Tieto typy amidov sa často označujú ako sekundárne a terciárne amidy, ktoré IUPAC neodporúča.
N-fenyl-substituované amidy majú vo svojom názve príponu -anilid. Poloha substituentov v anilínovom zvyšku je označená číslami s prvočíslami:
Okrem toho sa zachovali polosystematické názvy, v ktorých sa spája prípona -amid so základom latinského názvu karboxylovej kyseliny (formamid, acetamid), ako aj niektoré triviálne názvy ako "anilidy" (acylované anilíny) alebo "toluididy" (acylované toluidíny).
Amidy sú kryštalické látky s relatívne vysokými a zreteľnými teplotami topenia, čo umožňuje použitie niektorých z nich ako derivátov na identifikáciu karboxylových kyselín. V ojedinelých prípadoch sú to kvapaliny, napríklad amidy kyseliny mravčej - formamid a N,N-dimetylformamid - známe dipolárne aprotické rozpúšťadlá. Nižšie amidy sú vysoko rozpustné vo vode.
Amidy sú jeden z najodolnejších voči hydrolýze funkčné deriváty karboxylových kyselín, vďaka čomu sú v prírode široko rozšírené. Mnohé amidy sa používajú ako lieky. Asi storočie sa v lekárskej praxi používajú paracetamol a fenacetín, čo sú substituované amidy kyseliny octovej.
Štruktúra amidov. Elektrónová štruktúra amidovej skupiny je do značnej miery podobná štruktúre karboxylovej skupiny. Amidová skupina je p,π-konjugovaný systém, v ktorom je osamotený pár elektrónov atómu dusíka konjugovaný s elektrónmi väzby C=O π. Delokalizácia elektrónovej hustoty v amidovej skupine môže byť reprezentovaná dvoma rezonančnými štruktúrami:
V dôsledku konjugácie má väzba C-N v amidoch čiastočne bilinkované charakter, jej dĺžka je výrazne menšia ako dĺžka jednoduchej väzby v amínoch, zatiaľ čo väzba C=O je o niečo dlhšia ako väzba C=O v aldehydoch a ketónoch. Amidová skupina v dôsledku konjugácie má plochú konfiguráciu . Nižšie sú uvedené geometrické parametre molekuly N-substituovaného amidu, stanovené pomocou rôntgenovej difrakčnej analýzy:

Dôležitým dôsledkom čiastočne dvojito viazanej povahy väzby C-N je pomerne vysoká energetická bariéra pre rotáciu okolo tejto väzby; napríklad pre dimetylformamid je to 88 kJ/mol. Z tohto dôvodu môžu amidy s rôznymi substituentmi na atóme dusíka existovať vo forme n-diastereomérov. N-substituované amidy existujú prevažne ako Z-izoméry:

V prípade N,N-disubstituovaných amidov závisí pomer E- a Z-izomérov od objemu radikálov pripojených k atómu dusíka. Stereoizoméry amidov sú konfiguračne nestabilné, ich existencia bola preukázaná najmä fyzikálno-chemickými metódami, v individuálnej forme boli izolované len ojedinele. Je to spôsobené tým, že bariéra rotácie pre amidy stále nie je taká vysoká ako pre alkény, pre ktoré je to 165 kJ/mol.
Acidobázické vlastnosti. Amidy majú slabé kyslé aj zásadité vlastnosti . Zásaditosť amidov leží v rozmedzí hodnôt Pk BH + od -0,3 do -3,5. Dôvodom zníženej zásaditosti aminoskupiny v amidoch je konjugácia osamelého páru elektrónov atómu dusíka s karbonylovou skupinou. Pri interakcii so silnými kyselinami sú amidy protónované na atóme kyslíka v zriedených aj koncentrovaných roztokoch kyselín. Tento druh interakcie je základom kyslá katalýza pri reakciách hydrolýzy amidov:
Vykazujú sa nesubstituované a N-substituované amidy slabé vlastnosti NH-kyselín , porovnateľné s kyslosťou alkoholov a odstraňujú protón len pri reakciách so silnými zásadami.
Acidobázické interakcie sú základom tvorby amidov intermolekulárnych spolupracovníkov , ktorého existencia vysvetľuje vysoké teploty topenia a varu amidov. Je možná existencia dvoch typov asociátov: lineárne polyméry a cyklické diméry. Prevaha jedného alebo druhého typu je určená štruktúrou amidu. Napríklad N-metylacetamid, pre ktorý je výhodná Z-konfigurácia, tvorí lineárny asociát a laktámy s pevne fixovanou E-konfiguráciou tvoria diméry:

N,N-disubstituované amidy tvoria diméry v dôsledku interakcie dipól-dipól 2 polárnych molekúl:

Acylačné reakcie. V dôsledku prítomnosti silnej elektrón-donornej aminoskupiny v konjugovanom amidovom systéme je elektrofilita karbonylového uhlíkového atómu, a teda reaktivita amidov pri acylačných reakciách, veľmi nízka. Nízka acylačná schopnosť amidov sa vysvetľuje aj skutočnosťou, že amidový ión NH 2 - je zlou odstupujúcou skupinou. Z acylačných reakcií je dôležitá hydrolýza amidov, ktorá sa môže uskutočňovať v kyslom a alkalickom prostredí. Amidy sa hydrolyzujú oveľa ťažšie ako iné funkčné deriváty karboxylových kyselín. Hydrolýza amidov sa uskutočňuje za prísnejších podmienok v porovnaní s hydrolýzou esterov.
Kyslá hydrolýza amidy - nezvratné reakcia vedúca k tvorbe karboxylovej kyseliny a amónnej soli:
Vo väčšine prípadov kyslá hydrolýza amidov prebieha podľa mechanizmu acylácia bimolekulárnej kyseliny A AC 2 t.j. podobný mechanizmu kyslej hydrolýzy esterov. Nevratnosť reakcie je spôsobená skutočnosťou, že amoniak alebo amín sa v kyslom prostredí premieňa na amónny ión, ktorý nemá nukleofilné vlastnosti:

Alkalická hydrolýza To isté nezvratné reakcia; v dôsledku toho sa vytvorí soľ karboxylovej kyseliny a amoniak alebo amín:
Alkalická hydrolýza amidov, podobne ako hydrolýza esterov, prebieha podľa tetraedrický mechanizmus IN AC 2 . Reakcia začína pridaním hydroxidového iónu (nukleofilu) k elektrofilnému atómu uhlíka amidovej skupiny. Výsledný anión (I) je protónovaný na atóme dusíka a potom sa v bipolárnom ióne (II) vytvorí dobrá odstupujúca skupina - molekula amoniaku alebo amínu. Predpokladá sa, že pomalým štádiom je rozklad tetraedrického medziproduktu (II).
V prípade anilidov a iných amidov so substituentmi priťahujúcimi elektróny na atóme dusíka môže rozklad tetraedrického medziproduktu (I) prebiehať tvorbou dianiónu (II):

Trávenie kyseliny dusičnej. Pri interakcii s kyselinou dusitou a inými nitrozačnými činidlami sa amidy premieňajú na zodpovedajúce karboxylové kyseliny s výťažkom až 90 %:
Dehydratácia. Nesubstituované amidy sa vplyvom oxidu fosforečného a niektorých ďalších činidiel (POC1 3, PC1 5, SOCl 2) premieňajú na nitrily:
![]()
47. Karboxylové kyseliny: halogenácia podľa Gell-Volhard-Zelinsky, využitie reakcie na syntézu a -hydroxy a a -aminokyseliny.
Halogenácia alifatických karboxylových kyselín.
Alifatické karboxylové kyseliny sú halogenované do polohy α chlórom alebo brómom v prítomnosti katalytických množstiev červený fosfor alebo halogenidy fosforu (Gell-Volhard-Zelinsky reakcia ). Napríklad, keď sa kyselina hexánová brómuje v prítomnosti červeného fosforu alebo chloridu fosforitého, vo vysokom výťažku vzniká kyselina 2-brómhexánová, napríklad:
Nie je to samotná karboxylová kyselina, ktorá podlieha bromácii, ale chlorid kyseliny vytvorený z nej in situ. Chlorid kyseliny má silnejšie vlastnosti CH-kyseliny ako karboxylová kyselina a ľahšie tvorí enolovú formu.
Enol (I) pridáva bróm za vzniku halogénového derivátu (II), ktorý následne eliminuje halogenovodík a mení sa na a-halogénovaný halogenid kyseliny (III). V poslednom štádiu sa regeneruje halogenid kyseliny nesubstituovanej karboxylovej kyseliny.

Z výsledných a-halogénom substituovaných kyselín sa pomocou nukleofilných substitučných reakcií syntetizujú ďalšie heterofunkčné kyseliny.
Hydrolýza esterov a všetkých ostatných derivátov kyselín vyžaduje kyslú alebo alkalickú katalýzu. Kyslou hydrolýzou vznikajú karboxylové kyseliny a alkoholy (reverzná esterifikačná reakcia), alkalickou hydrolýzou vznikajú soli karboxylových kyselín a alkoholov.
Kyslá hydrolýza esterov:
Mechanizmus SN, nukleofil - H 2 O, alkoxyskupina je nahradená hydroxyl.
Alkalická hydrolýza esterov: reakcia prebieha v dvoch stupňoch s 2 mólmi zásady, výsledná kyselina sa premení na soľ.
Mechanizmus S N, Nu = - OH
Tvorba zlúčenín podobných soliam Amidy sú neutrálne látky, pretože základné vlastnosti amoniaku sú oslabené nahradením atómu vodíka v ňom kyslým zvyškom. Preto skupina NH2 v amidoch na rozdiel od amínov len ťažko tvorí óniový katión. So silnými kyselinami však amidy poskytujú soli, napríklad Cl, ktoré sa vodou ľahko rozkladajú. Na druhej strane, vodík skupiny NH2 v amidoch sa ľahšie nahrádza kovmi ako v amoniaku a amínoch. Acetamid napríklad ľahko rozpúšťa oxid ortutnatý, čím vzniká zlúčenina (CH3CONH)2Hg.
Je však možné, že počas tvorby kovových derivátov dôjde k izomerizácii amidu a výsledná zlúčenina má izomérnu (tautomerickú) štruktúru soli imidokyseliny
t.j. existuje analógia so soľami kyseliny kyanovodíkovej.
2. Pôsobenie kyseliny dusnej Amidy reagujú s kyselinou dusitou, podobne ako primárne amíny, za vzniku karboxylových kyselín a uvoľnenia dusíka:
3. Zmydelnenie Pri varení s minerálnymi kyselinami a zásadami pridávajú amidy vodu za vzniku karboxylovej kyseliny a amoniaku:
4. Pôsobenie alkylhalogenidov. Pôsobením alkylhalogenidov na amidy alebo ich kovové deriváty vznikajú N-substituované amidy:
5. Účinok chloridu fosforečného. Pôsobením chloridu fosforečného na amidy vzniká chlóramidy
ľahko sa rozkladá na kyselinu chlorovodíkovú a imid chloridy
Ten s amoniakom môže produkovať soli amidíny;
6. Konverzia na amíny. Intenzívnou redukciou amidov možno získať primárne amíny s rovnakým počtom atómov uhlíka:
7. Hoffmannova reakcia. Keď sú amidy vystavené pôsobeniu hypohalogenitu alebo brómu a alkálie, tvoria sa amíny a atóm uhlíka karbonylovej skupiny sa odštiepi vo forme C02 (A. Hoffman). Priebeh reakcie možno znázorniť nasledovne:
Vo vzdelávacích príručkách sa stále často nachádza iná interpretácia mechanizmu tejto reakcie:
Tento priebeh reakcie je však menej pravdepodobný, pretože sa vytvorí fragment
s atómom dusíka nesúcim dva voľné elektrónové páry je nepravdepodobné.
Tomuto mechanizmu odporuje najmä skutočnosť, že ak je radikál R opticky aktívny, potom v dôsledku reakcie neracemizuje. Medzitým by aj prchavá existencia voľného radikálu R – : viedla k strate optickej aktivity.
Chemické vlastnosti. Nitroskupina je jednou z najviac silné skupiny priťahujúce elektróny a je schopný efektívne delokalizovať negatív. poplatok. V aromatickom spoj. v dôsledku indukčného a najmä mezomérneho efektu ovplyvňuje rozloženie hustoty elektrónov: jadro sa stáva čiastočne kladným. náboj, ktorý je lokalizovaný Ch. arr. v orto a para polohách; Hammettove konštanty pre skupinu NO2 s m 0,71, s n 0,778, s + n 0,740, s - n 1,25. Zavedenie skupiny N02 teda prudko zvyšuje reakciu. schopnosť org. spoj. vo vzťahu k nukleofilným činidlám a sťažuje prácu s elektrof. činidlá. To určuje široké použitie nitrozlúčenín v org. syntéza: skupina NO 2 sa zavedie do požadovanej polohy molekuly org. spojenie, vykonať rozklad. ptóny spojené spravidla so zmenou uhlíkového skeletu a potom transformované na inú funkciu alebo odstránené. V aromatickom V niektorých prípadoch sa často používa kratšia schéma: nitrácia-transformácia skupiny NO 2 .
Tvorba nitrónových zlúčenín v sérii aromatických nitrozlúčenín je spojená s izomerizáciou benzénového kruhu na chinoidnú formu; napríklad nitrobenzén sa tvorí s konc. H 2 SO 4 farebný produkt podobný soli typu I, o-nitrotoluén, vykazuje fotochromizmus v dôsledku intramol. prenos protónov za vzniku jasne modrého derivátu O:
Pri pôsobení zásad na primárne a sekundárne nitrozlúčeniny vznikajú soli nitrozlúčenín; okolité anióny solí v roztokoch s elektrofilmi sú schopné produkovať O- aj C-deriváty. Keď sa teda soli nitrozlúčenín alkylujú alkylhalogenidmi, trialkylchlórsilánmi alebo R30 + BF-4, tvoria sa O-alkylačné produkty. Najnovšie m.b. získava sa tiež pôsobením diazometánu alebo N,O-bis-(trimetylsilyl)acetamidu na nitroalkány s pKa< 3 или нитроновые к-ты, напр.:
Acyklický alkylestery nitrónových kyselín sú tepelne nestabilné a rozpadajú sa intramol. mechanizmus:
R-ts a s r a r s v asi m so spojeniami a S-N. Primárne a sekundárne nitrozlúčeniny pri zahrievaní. s minerálom K-tami je prítomný. alkohol alebo vodný roztok alkalických zlúčenín tvoria karbonylové zlúčeniny. (pozri Nave reakcia). R-tion prechádza cez medzery. tvorba nitrónových zlúčenín:
Ako počiatočné spojenie Môžu sa použiť silylnitrón étery. Účinok silných zlúčenín na alifatické nitrozlúčeniny môže viesť k hydroxámovým zlúčeninám, napríklad:
Existuje mnoho známych spôsobov redukcie nitrozlúčenín na amíny. Široko používané sú železné piliny, Sn a Zn. súprava; s katalytickým pri hydrogenácii sa ako katalyzátory používajú Ni-Raney, Pd/C alebo Pd/PbCO 3 a iné Alifatické nitrozlúčeniny sa v prítomnosti ľahko redukujú na amíny LiAlH 4 a NaBH 4 . Pd, Na a Al amalgámy, pri zahrievaní. s hydrazínom nad Pd/C; pre aromatické nitrozlúčeniny sa niekedy používa TlCl 3, CrCl 2 a SnCl 2, aromatické. polynitrozlúčeniny sú selektívne redukované na nitramíny hydrosulfidom sodným v CH30H. Existujú spôsoby, ako si vybrať. redukcia skupiny NO 2 v polyfunkčných nitrozlúčeninách bez ovplyvnenia iných funkcií.
Keď P(III) pôsobí na aromatické nitrozlúčeniny, vzniká sekvencia. deoxygenácia skupiny NO 2 za vzniku vysoko reaktívnych nitrénov. Roztok sa používa na syntézu kondenzátora. heterocykly, napríklad:
R-ts a zachovanie skupiny N02. Alifatické nitrozlúčeniny obsahujúce a-H atóm sa ľahko alkylujú a acylujú, zvyčajne tvoria O-deriváty. Avšak vzájomný mod. dilítiové soli primárnych nitrozlúčenín s alkylhalogenidmi, anhydridmi alebo halogenidmi karboxylových kyselín vedú k C-alkylačným alebo C-acylačným produktom, napr.
Sú známe príklady intramol. C-alkylácia, napr.
Primárne a sekundárne nitrozlúčeniny reagujú s alifatickými zlúčeninami. amíny a CH20 za vzniku p-aminoderivátov (Mannichov roztok); v roztoku môžete použiť vopred pripravené metylolové deriváty nitrozlúčenín alebo aminozlúčenín:
Nitrometán a nitroetán môžu kondenzovať s dvoma molekulami metylolamínu a vyššie nitroalkány iba s jednou. Pri určitých pomeroch činidiel môže roztok viesť k heterocyklu. spojenie, napríklad: pri interakcii vzniká primárny nitroalkán s dvoma ekvivalentmi primárneho amínu a nadbytkom formaldehydu. Formy V, ak sa činidlá odoberajú v pomere 1:1:3-kom. Formuláre VI.
Aromatické nitrozlúčeniny ľahko vstupujú do nukleofilných roztokov. substitúcie a oveľa ťažšie - v obvode elektrof. substitúcia; v tomto prípade je nukleofil nasmerovaný do orto- a po-pozícií a elektrofil je nasmerovaný do meta-polohy k N02 skupine. Elektrická konštantná rýchlosť nitrácia nitrobenzénu je o 5 až 7 rádov nižšia ako nitrácia benzénu; tým vzniká m-dinitrobenzén.
Pri karboxylácii primárnych nitroalkánov pôsobením CH 3 OMgOCOOCH 3 vznikajú a-nitrouhlíkové zlúčeniny alebo ich estery.
Pri úprave solí mononitrozlúčenín C(NO 2) 4 s Ag alebo dusitanmi alkalických kovov alebo pri pôsobení dusitanov na a-halogénnitroalkány v alkalickom prostredí (Ter Meerov roztok) vznikajú hemodinitrozlúčeniny Elektrolýza tzv. a-halogénnitroalkány v aprotických roztokoch, ako aj spracovanie nitrozlúčenín Cl2 v alkalickom prostredí alebo elektrooxidácia solí nitrozlúčenín vedú k vic-dinitrozlúčeninám:
Nitro skupina nevykresľuje bytosti. vplyv na radikálovú alkyláciu alebo aryláciu aromatických. spoj.; r-tion vedie k zákl. na orto- a para-substituované produkty.
Na redukciu nitrozlúčenín bez ovplyvnenia skupiny NO 2 použite NaBH 4, LiAlH 4 pri nízkych teplotách alebo roztok diboránu v THF, napríklad:
Aromatické di- a trinitrozlúčeniny, najmä 1,3,5-trinitrobenzén, tvoria stabilné, pestrofarebné kryštalické zlúčeniny. hovoria komplexy s aromatickými zlúčeniny donorov elektrónov (amíny, fenoly atď.). Komplexy s kyselinou pikrovou sa používajú na izoláciu a čistenie aromatických zlúčenín. uhľovodíky. Interakcia di- a trinitrobenzénov so silnými zásadami (HO -, RO -, N - 3, RSO - 2, CN -, alifatické amíny) vedie k tvorbe Meisen-Haimerových komplexov, ktoré sú izolované vo forme farebných solí alkalických kovov.
Vhodné oxidačné činidlá pre tieto reakcie sú kyselina chrómová alebo dusičná, zmes chrómu, oxid manganičitý alebo oxid seléničitý.
Pri oxidácii kyselinou chrómovou sa alkohol nukleofilne naviaže na kyselinu chrómovú, pri ktorej sa odštiepi voda a vznikne ester kyseliny chrómovej (ide o prvý stupeň reakcie, je to podobné ako pri vzniku esterov karboxylových kyselín, porovnaj oddiel E, 7.1.5.1). V druhom stupni, ktorý pravdepodobne prechádza cyklickým prechodným stavom, a-vodík alkoholu prechádza na chromátový zvyšok a kov prechádza zo šesťmocného stavu do štvormocného:
| n-CH30> P-terc-C4H9> P-CH3> P-Cl> P- NIE 2 | (G.6.20) |
Keď sú primárne alkoholy oxidované, výsledný aldehyd musí byť chránený pred ďalšou oxidáciou na karboxylovú kyselinu. Je napríklad možné neustále oddestilovať aldehyd z reakčnej zmesi: je to celkom uskutočniteľné, pretože teplota varu aldehydu je zvyčajne nižšia ako teplota varu zodpovedajúceho alkoholu. Napriek tomu výťažok aldehydov počas oxidácie dichrómanom zriedka prekračuje 60 %. Je pozoruhodné, že keď sa reakcia vykonáva správne, viacnásobné väzby uhlík-uhlík sú takmer nedotknuté.
Aldehydy sa tiež tvoria, keď sa alkoholy zahrievajú s vodným neutrálnym roztokom dichrómanu, ale dobré výťažky poskytujú iba benzylalkoholy.
Vyššie výťažky aldehydov možno získať oxidáciou primárnych alkoholov drhne-butylchróman (v petroléteri, benzéne alebo tetrachlórmetáne) alebo oxid manganičitý (v acetóne, petroléteri, tetrachlórmetáne alebo zriedenej kyseline sírovej). Tieto činidlá tiež umožňujú získať nenasýtené a aromatické aldehydy v dobrých výťažkoch.
Oxidácia sekundárnych alkoholov na ketóny je ešte jednoduchšia ako oxidácia primárnych alkoholov. Výťažky sú tu vyššie, pretože po prvé, reaktivita sekundárnych alkoholov je vyššia ako reaktivita primárnych alkoholov a po druhé, výsledné ketóny sú oveľa odolnejšie voči oxidácii v porovnaní s aldehydmi. Zo steroidov a terpénov sa osvedčila oxidácia sekundárnych alkoholov komplexom kyseliny chrómovej a pyridínu, ako aj anhydridu kyseliny chrómovej v dimetylformamide. Anhydrid kyseliny chrómovej v acetóne je tiež dobré oxidačné činidlo; môže sa použiť na oxidáciu nenasýtených sekundárnych alkoholov bez ovplyvnenia viacnásobnej väzby uhlík-uhlík.
Novou metódou, vhodnou aj pre stéricky bránené alkoholy, je oxidácia dimetylsulfoxidom v acetanhydride.
Podľa postupu uvedeného nižšie sa reakcia uskutočňuje v dvojfázovom systéme. Výsledné ketóny sa extrahujú organickým rozpúšťadlom a sú tak chránené pred ďalšou oxidáciou.
Disacharidy– uhľohydráty, ktorých molekuly pozostávajú z dvoch monosacharidových zvyškov, ktoré sú navzájom spojené interakciou dvoch hydroxylových skupín.
Počas tvorby disacharidovej molekuly sa eliminuje jedna molekula vody:
alebo pre sacharózu:
Preto je molekulový vzorec disacharidov C12H22O11.
K tvorbe sacharózy dochádza v rastlinných bunkách pod vplyvom enzýmov. Chemici však našli spôsob, ako uskutočniť mnohé reakcie, ktoré sú súčasťou procesov, ktoré sa vyskytujú v živej prírode. V roku 1953 francúzsky chemik R. Lemieux prvýkrát uskutočnil syntézu sacharózy, ktorú súčasníci nazvali „dobytím Everestu organickej chémie“.
V priemysle sa sacharóza získava zo šťavy z cukrovej trstiny (obsah 14 – 16 %), cukrovej repy (16 – 21 %), ako aj niektorých ďalších rastlín, ako je kanadský javor alebo hruška hlinená.
Každý vie, že sacharóza je kryštalická látka, ktorá má sladkú chuť a je vysoko rozpustná vo vode.
Šťava z cukrovej trstiny obsahuje sacharid sacharózu, bežne nazývanú cukor.
S výrobou cukru z repy je úzko spojené meno nemeckého chemika a hutníka A. Marggrafa. Bol jedným z prvých výskumníkov, ktorí pri svojom chemickom výskume použili mikroskop, pomocou ktorého v roku 1747 objavil kryštály cukru v repnej šťave.
Laktóza – kryštalická mliečny cukor, sa získaval z mlieka cicavcov už v 17. storočí. Laktóza je menej sladký disacharid ako sacharóza.
Teraz sa zoznámime so sacharidmi, ktoré majú zložitejšiu štruktúru - polysacharidy.
Polysacharidy– uhľohydráty s vysokou molekulovou hmotnosťou, ktorých molekuly pozostávajú z mnohých monosacharidov.
V zjednodušenej forme môže byť všeobecná schéma prezentovaná takto:
Teraz si porovnajme štruktúru a vlastnosti škrobu a celulózy – najvýznamnejších predstaviteľov polysacharidov.
Štruktúrnou jednotkou polymérnych reťazcov týchto polysacharidov, ktorých vzorec je (C6H10O5)n, sú glukózové zvyšky. Aby ste mohli zapísať zloženie štruktúrnej jednotky (C 6 H 10 O 5), musíte od vzorca glukózy odpočítať molekulu vody.
Celulóza a škrob sú rastlinného pôvodu. Vznikajú z molekúl glukózy v dôsledku polykondenzácie.
Rovnicu pre polykondenzačnú reakciu, ako aj jej inverzný proces hydrolýzy pre polysacharidy, možno podmienečne zapísať takto:
Molekuly škrobu môžu mať lineárny aj rozvetvený typ štruktúry, molekuly celulózy môžu mať len lineárnu štruktúru.
Pri interakcii s jódom škrob, na rozdiel od celulózy, dáva modrú farbu.
Tieto polysacharidy majú tiež rôzne funkcie v rastlinných bunkách. Škrob slúži ako rezervná živina, celulóza plní štrukturálnu, stavebnú funkciu. Bunkové steny rastlín sú vyrobené z celulózy.
CANNIZZAROREAKCIA, oxidačno-redukcia disproporcionácia aldehydov pod vplyvom alkálií s tvorbou primárnych alkoholov a uhličitých kyselín, napr.
Aldehyd sa spracuje konc. vodný alebo vodno-alkoholický roztok alkálie pri ochladení alebo miernom zahriatí.katalyzátory – rozklad. kovy (napr. Ag, Ni, Co, Cu) a ich oxidy. Do roztoku vstupujú aldehydy, ktoré neobsahujú atóm H v polohe a ku karbonylovej skupine. Inak nie je výhodná Cannizzarova reakcia, ale skôr aldolová kondenzácia. Substituenty priťahujúce elektróny v aromatickom kruhu. aldehydy tento proces urýchľujú a elektrónové darcovské spomaľujú. Benzaldehydy so substituentmi v orto polohách nereagujú v Cannizzaro; o- a p-hydroxybenzaldehydy reagujú iba v prítomnosti. Ag. V kapitole je použitá reakcia využívajúca dva rôzne aldehydy (tzv. krížová Cannizzarova reakcia). arr. na získanie primárnych alkoholov z aromátov vo vysokom výťažku. aldehydy. Formaldehyd sa zvyčajne používa ako redukčné činidlo:
ArCHO + CH20: ArCH2OH + HCOOH
Počas syntézy polyhydroxymetylovaných zlúčenín. formaldehyd sa v prvom stupni zúčastňuje aldolovej kondenzácie a potom ako redukčné činidlo v Cannizzarovej krížovej reakcii:
Cannizzarov navrhovaný mechanizmus pre homogénnu reakciu. prostredie zahŕňa stupeň prenosu hydridu
Pre aromatické aldehydov, nemožno vylúčiť možnosť účasti na Cannizzarovej reakcii radikálových aniónov vytvorených v dôsledku prenosu jedného elektrónu. S intramolom nastáva reakcia podobná Cannizzarovej reakcii. disproporcionácia a-ketoaldehydov v prítomnosti. alkálie (Cannizzaro prešmyk):
Cannizzarova reakcia sa používa na priemyselné účely. syntéza pentaerytritolu, prípravná výroba alkoholov, uhlíkatých zlúčenín atď. Proces objavil v roku 1853 S. Cannizzaro.
Pyrol, furán a tiofén sú päťčlenné heterocyklické zlúčeniny s jedným heteroatómom.
Číslovanie atómov v heterocykle začína heteroatómom a ide proti smeru hodinových ručičiek. Pozície 2 a 5 sa nazývajú a-pozície, 3 a 4 sa nazývajú b-pozície.
Podľa formálnych charakteristík sú tieto zlúčeniny klasifikované ako aromatické, pretože sú to konjugované cyklické p-systémy, ktoré zahŕňajú 6p elektróny - 4 elektróny diénového systému - a pár heteroatómových elektrónov. Cyklus je takmer plochý, čo znamená, že hybridizačný stav heteroatómu je blízky sp 2.
Nižšie sú rezonančné štruktúry ilustrujúce delokalizáciu elektrónov heteroatómu pozdĺž heterocyklického kruhu s použitím furánu ako príkladu.
Vyššie uvedené rezonančné štruktúry ukazujú, že heteroatóm (v tomto prípade atóm kyslíka) v dôsledku mezomérnej interakcie s diénovým π-systémom prenáša elektrónovú hustotu do kruhu, v dôsledku čoho sa na kruhu objaví určitý negatívny náboj. atómov uhlíka v heterocykle, a teda kladný náboj na náboji atómu kyslíka. Atóm kyslíka má samozrejme okrem pozitívneho mezomérneho efektu aj negatívny indukčný efekt. Jeho prejav vo vlastnostiach uvažovaných zlúčenín je však menej výrazný, a preto sa päťčlenné heterocykly s jedným heteroatómom zaraďujú medzi aromatické heterocyklické zlúčeniny s prebytkom p. Rezonancia vedie k určitému vyrovnaniu dĺžok väzieb v heterocykle, čo tiež naznačuje určitú aromatickosť systému.
DEFINÍCIA
Zlúčeniny organickej povahy, ktoré sú derivátmi karboxylových kyselín, vznikajú pri ich interakcii s alkoholmi:
Všeobecný štruktúrny vzorec esterov:

kde R a R' sú uhľovodíkové radikály.
Hydrolýza esterov
Jednou z najcharakteristickejších schopností esterov (okrem esterifikácie) je ich hydrolýza - štiepenie vplyvom vody. Iným spôsobom sa hydrolýza esterov nazýva saponifikácia. Na rozdiel od hydrolýzy solí je v tomto prípade prakticky nevratná. Rozlišuje sa alkalická a kyslá hydrolýza esterov. V oboch prípadoch sa tvorí alkohol a kyselina:
a) kyslá hydrolýza

b) alkalická hydrolýza

Príklady riešenia problémov
PRÍKLAD 1
| Cvičenie | Určte hmotnosť kyseliny octovej, ktorá sa môže získať počas zmydelňovacej reakcie etylacetátu s hmotnosťou 180 g. |
| Riešenie | Napíšme reakčnú rovnicu pre hydrolýzu etylesteru kyseliny octovej pomocou hrubého vzorca: C 4 H 8 O 2 + H 2 O ↔ CH 3 COOH + C 2 H 5 OH. Vypočítajme množstvo etylacetátovej látky (mólová hmotnosť - 88 g/mol) pomocou hodnoty hmotnosti z problémových podmienok: a(C4H802) = m (C4H802)/M (C4H802) = 180/88 = 2 mol. Podľa reakčnej rovnice je počet mólov etylacetátu a kyseliny octovej rovnaký: υ (C4H802) = υ (CH3COOH) = 2 mol. Potom môžete určiť hmotnosť kyseliny octovej (molárna hmotnosť - 60 g / mol): m(CH3COOH) = a(CH3COOH) x M (CH3COOH) = 2 x 60 = 120 g. |
| Odpoveď | Hmotnosť kyseliny octovej je 120 gramov. |
Hydrolýza esterov je katalyzovaná kyselinami aj zásadami. Kyslá hydrolýza esterov sa zvyčajne uskutočňuje zahrievaním s kyselinou chlorovodíkovou alebo sírovou vo vodnom alebo vodno-alkoholickom prostredí. V organickej syntéze sa pre mono- a dialkylsubstituované estery kyseliny malónovej najčastejšie využíva kyslá hydrolýza esterov (kapitola 17). Mono- a disubstituované deriváty esteru kyseliny malónovej podliehajú hydrolýze, keď sa varia s koncentrovanou kyselinou chlorovodíkovou, po ktorej nasleduje dekarboxylácia.
Na hydrolýzu katalyzovanú zásadou sa zvyčajne používa vodný alebo vodno-alkoholický roztok NaOH alebo KOH. Najlepšie výsledky sa dosiahnu použitím riedkej suspenzie hydroxidu draselného v DMSO obsahujúcej malé množstvo vody.

Posledný spôsob je výhodný na zmydelnenie bránených esterov kyselín; ďalšou modifikáciou tohto spôsobu je alkalická hydrolýza bránených esterov v prítomnosti 18-crown-6-polyesteru:

Na preparatívne účely má zásadou katalyzovaná hydrolýza množstvo zrejmých výhod oproti kyslej hydrolýze. Rýchlosť zásaditej hydrolýzy esterov je zvyčajne tisíckrát vyššia ako pri kyslej katalýze. Hydrolýza v kyslom prostredí je reverzibilný proces, na rozdiel od hydrolýzy v prítomnosti zásady, ktorá je nevratná.
18.8.2.A. Mechanizmy hydrolýzy esterov
Hydrolýza esterov čistou vodou je vo väčšine prípadov reverzibilná reakcia, ktorá vedie k rovnovážnej zmesi karboxylovej kyseliny a materského esteru:

Táto reakcia je výrazne zrýchlená v kyslom a alkalickom prostredí, čo je spojené s acidobázickou katalýzou (kapitola 3).
Podľa K. Ingolda sú mechanizmy hydrolýzy esterov klasifikované podľa nasledujúcich kritérií:
(1) Typ katalýzy: kyslá (symbol A) alebo zásaditá (symbol B);
(2) Typ štiepenia označujúci, ktorá z dvoch C-O -väzieb v esteri sa štiepi v dôsledku reakcie: acylový kyslík (AC index) alebo alkylový kyslík (AL index):

(3) Molekularita reakcie (1 alebo 2).
Z týchto troch kritérií možno vytvoriť osem rôznych kombinácií, ktoré sú znázornené v diagrame 18.1.

Toto sú najbežnejšie mechanizmy. Alkalické zmydelnenie patrí takmer vždy k typu B AC 2. Kyslá hydrolýza (ako aj esterifikácia) má vo väčšine prípadov mechanizmus A AC 2.

Mechanizmus A AC 1 sa zvyčajne pozoruje len v silne kyslých roztokoch (napríklad v koncentrovanej H 2 SO 4) a najmä často pri stéricky bránených esteroch aromatických kyselín.
Mechanizmus AC 1 je stále neznámy.

Mechanizmus B AL 2 bol zistený len v prípade výnimočne silných priestorovo tienených acylových skupín a neutrálnej hydrolýzy β-laktónov. Mechanizmus A AL 2 je stále neznámy.

Podľa mechanizmu A AL 1 terciárne alkylestery zvyčajne reagujú v neutrálnom alebo kyslom prostredí. Rovnaké substráty za podobných podmienok môžu reagovať podľa mechanizmu B AL 1, avšak pri prechode do mierne alkalickejšieho prostredia je mechanizmus B AL 1 okamžite nahradený mechanizmom B AC 2.
Ako je zrejmé zo schémy 18.1, reakcie katalyzované kyselinami sú reverzibilné a z princípu mikroskopickej reverzibility (kapitola 2) vyplýva, že podobnými mechanizmami prebieha aj esterifikácia katalyzovaná kyselinami. Pri zásaditej katalýze sa však rovnováha posúva smerom k hydrolýze (zmydelneniu), pretože sa rovnováha posúva v dôsledku ionizácie karboxylovej kyseliny. Podľa vyššie uvedenej schémy sú v prípade mechanizmu A AC1 skupiny COOR a COOH protónované na alkoxy alebo hydroxylovom kyslíkovom atóme. Všeobecne povedané, z hľadiska termodynamiky je výhodnejšia protonácia karbonylového kyslíka, skupiny C=O, pretože v tomto prípade môže byť kladný náboj delokalizovaný medzi oboma atómami kyslíka:

Napriek tomu roztok v malom množstve obsahuje aj tautomérny katión - nevyhnutný medziprodukt v mechanizme A AC 1. Oba mechanizmy B1 (z ktorých B AC 1 nie je známy) v skutočnosti vôbec nie sú katalytické, pretože na začiatku dochádza k disociácii vzniká neutrálny ester.
Z ôsmich Ingoldových mechanizmov bolo experimentálne dokázaných iba šesť.