Iron intake and hemoglobin synthesis. Anemias caused by disorders of hemoglobin synthesis and iron metabolism. Lipolysis and lipogenesis. Meaning. Dependence of lipogenesis on the nutritional rhythm and food composition. Regulation of lipolysis and lipogenesis. Transport and use
The synthesis of the pyrrole complex in the body proceeds from low molecular weight precursors de novo. Sources of iron are foods and iron, which is released during the breakdown of red blood cells.
Heme synthesis.
Istage... Glycine and succinyl-CoA are involved. 5-aminolevulinate synthase- a key, allosteric enzyme for the synthesis of tetrapyrroles. Coenzyme - pyridoxal phosphate. Induced by steroids and inhibited by feedback by the end product, heme. Formed 5-aminolevulinic acid(-ALK).
IIstage... Education porphobilinogen PBG. Enzyme porphobilinogen synthase inhibited by the end products of the synthesis.
IIIstage... Multi-stage. Tetrapyrrole complex is synthesized from 4 porphobilinogen molecules protoporphyrinIX.
IVstage... Protoporphyrin IX binds iron with the participation ferrochelatases (hemsynthases), and formed heme... The source of iron is ferritin. Vitamin B 12 and copper ions are involved in the synthesis of heme.
Protein part the hemoglobin molecule is synthesized in the same way as all other proteins. The synthesis of hemoglobin polypeptide chains occurs only in the presence of heme.
2.7. Nucleoprotein exchange
The collapse of the NK. Under the influence of stomach enzymes, partly hydrochloric acid, food nucleoproteins break down into polypeptides and NK. The breakdown of NK occurs in the small intestine in a hydrolytic way under the action of nuclease pancreatic juice. They belong to phosphodiesterases. Exists endonucleases and exonuclease, ribonuclease and deoxyribonuclease. The hydrolysis products are mononucleotides and oligonucleotides. Nucleases cleave NK molecules in tissues.
Decomposition of nucleoside phosphates. The first stage is the elimination of the phosphoric acid residue. At the second stage, the residual caribose is transferred from the nucleoside to phosphoric acid. This reaction is accelerating ribosyltransferases.
F-U-A F + U-A; U-A + F U-F + A
Breakdown of purine bases begins with deamination of those that have amino groups. Specific aminohydrolases are involved.
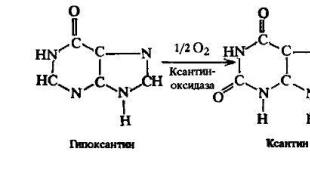
Adenine hypoxanthine; guanine xanthine
Hypoxanthine and xanthine oxidized to uric acid, the enzyme - xanthine oxidase.

Uric acid is produced mainly in the liver. It is the main product of purine nucleotide catabolism in humans. In the body, it forms 0.5-1 g per day, it is excreted through the kidneys. Chronic increase in the concentration of uric acid ( hyperuricemia) often leads to the development gout... Gouty crisis is associated with the deposition of sodium urate crystals in the joint. Hyperuricemia is usually hereditary.
Decomposition of pyrimidine bases also begins with deamination. Deaminated pyrimidine bases are being restored. Carbamic acid and-alanine are the end products of the decomposition of U and C. From T instead of-alanine, -aminoisobutyric acid is formed.
 …
…
Synthesis of pyrimidine nucleotides y, c, t
From CO 2 , gln, asp synthesized uridine monophosphoric acid... It serves as a precursor to cytidyl and thymidyl pyrimidine nucleotides.
The first reaction is carbamoyl phosphate formation under the action of carbamoyl phosphate synthetase II (glutamate-dependent, contained in the cytosol).
CO 2 + Glutamine + 2 ATP + H 2 O N 2 N CO ORO 3 N 2 + 2 ADP + H 3 RO 4 .
The carbamoyl phosphate then reacts with aspartate. As a result of a series of reactions, uridyl acid is formed.
Orotaciduria- excretion of large amounts of orotic acid in the urine. Known hereditary orotaciduria, in which up to 1.5 g of orotic acid is released per day, 1000 times more than normal. The disease is associated with a deficiency of an enzyme that catalyzes the formation and decarboxylation of orotidylic acid. Hereditary orotaciduria leads to the development of an irreversible sharp lag in mental and physical development; usually patients die in the first years of life. Orotic acid is not toxic; developmental disorders are the result of "pyrimidine hunger". Therefore, uridine is used to treat this disease.
Reduction - hydrogen donor - protein thioredoxin containing SH-groups;
Amination - the source of the amino group is gln;
- methylation source of the methyl group - methylene H 4 -folate.
When interacting with ATP, free pyrimidine nucleoside triphosphates are synthesized.

Erythrocyte synthesis- one of the most powerful processes of cell formation in the body. Normally, about 2 million red blood cells are formed every second, 173 billion a day, and 63 trillion a year. If we translate these values into mass, then about 140 g of erythrocytes are formed daily, every year - 51 kg, and the mass of erythrocytes formed in the body over 70 years is about 3.5 tons.
In an adult erythropoiesis occurs in the bone marrow of flat bones, while in the fetus islets of hematopoiesis are found in the liver and spleen (extramedullary hematopoiesis). In some pathological conditions (thalassemia, leukemia, etc.), foci of extramedullary hematopoiesis can be found in an adult.
One of the important elements of cell division is vitamin B required for DNA synthesis, being, in fact, a catalyst for this reaction. In the process of DNA synthesis, vitamin B₁₂ is not consumed, but cyclically enters into reactions as an active substance; as a result of this cycle, thymidine monophosphate is formed from uridine monophosphate. With a decrease in the level of vitamin B₁₂, uridine is poorly incorporated into the DNA molecule, which leads to numerous disorders, in particular, impaired maturation of blood cells.
Another factor that affects dividing cells is folic acid... She, as a coenzyme, in particular, is involved in the synthesis of purine and pyrimidine nucleotides.
General scheme of postembryonic hematopoiesis
Hematopoiesis(hematopoiesis) is a very dynamic, well-balanced, continuously renewing system. The single ancestor of hematopoiesis is the stem cell. According to modern concepts, this is a whole class of cells that are laid down in ontogenesis, the main property of which is the ability to give all the hematopoietic germs - erythrocytic, megakaryocytic, granulocytic (eosinophils, basophils, neutrophils), monocytic-macrophage, T-lymphocytic, B-lymphocytic.
As a result of several divisions, cells lose their ability to be universal ancestors and turn into pluripotent cells. Such, for example, is the precursor cell of myelopoiesis (erythrocytes, megakaryocytes, granulocytes). After a few more divisions, after universality, pluripotency also disappears, the cells become unipotent (ˮuniˮ is the only one), that is, they are capable of differentiation in only one direction.

The most dividing cells in the bone marrow are the precursor cells of myelopoiesis (see figure ⭡); as differentiation proceeds, the number of remaining divisions decreases, and morphologically distinguishable red blood cells gradually stop dividing.
Differentiation of erythroid cells
The erythroid cell line itself (erythron) begins with unipotent burst-forming cells, which are descendants of myelopoiesis progenitor cells. Burst-forming cells in tissue culture grow in small colonies resembling an explosion (burst). For their maturation, a special mediator is required - burst-promoter activity. This is a factor of influence of the microenvironment on maturing cells, a factor of intercellular interaction.
There are two populations of burst-forming cells: the first is regulated exclusively by burst-promoter activity, the second becomes sensitive to the effects of erythropoietin. In the second population begins hemoglobin synthesis continuing in erythropoietin-sensitive cells and in subsequent maturing cells.
At the stage of burst-forming cells, a fundamental change in cellular activity occurs - from division to the synthesis of hemoglobin. In subsequent cells, division is suspended (the last cell in this row capable of division is the polychromatophilic erythroblast), the nucleus decreases in absolute size and in relation to the volume of the cytoplasm, in which the synthesis of substances is taking place. At the last stage, the nucleus is removed from the cell, then the remnants of RNA disappear; they can still be found with a special stain in young erythrocytes - reticulocytes, but cannot be found in mature erythrocytes.
The scheme of the main stages of differentiation of erythroid cells is as follows:
pluripotent stem cell ⭢ burst-forming unit of the erythroid series (BFU-E) ⭢ colony-forming unit of the erythroid series (CFU-E) ⭢ erythroblast ⭢ pronormocyte ⭢ basophilic normocyte ⭢ polychromatic normocyte ⭢ orthochromatic (oxyphilic) erythroid normocyte.
Regulation of erythropoiesis
The processes of regulation of hematopoiesis are still insufficiently studied. The need to continuously maintain hematopoiesis, to adequately satisfy the body's needs in various specialized cells, to ensure the constancy and equilibrium of the internal environment (homeostasis) - all this presupposes the existence of complex regulatory mechanisms acting on the principle of feedback.
The most famous humoral factor in the regulation of erythropoiesis is the hormone erythropoietin... It is a stress factor that is synthesized in various cells and in various organs. Most of it is formed in the kidneys, however, even in their absence, erythropoietin is produced by the vascular endothelium and the liver. The level of erythropoietin is stable and changes upward with a sharp and profuse blood loss, acute hemolysis, when climbing mountains, with acute renal ischemia. Paradoxically, in chronic anemia, erythropoietin levels are usually normal, with the exception of aplastic anemia, where its level is consistently extremely high.
Along with erythropoietin, erythropoiesis inhibitors are also present in the blood. This is a large number of various substances, some of which can be attributed to medium-molecular toxins that accumulate as a result of pathological processes associated with their increased formation or impairment of their excretion.
In the early stages of differentiation, regulation in erythron is carried out mainly due to factors of the cellular microenvironment, and later - with a balance of the activity of erythropoietin and inhibitors of erythropoiesis. In acute situations, when it is necessary to quickly create a large number of new erythrocytes, the stress erythropoietin mechanism is triggered - a sharp predominance of erythropoietin activity over the activity of erythropoietic inhibitors. In pathological situations, on the contrary, inhibitory activity may prevail over erythropoietin, which leads to inhibition of erythropoiesis.
Hemoglobin synthesis
Hemoglobin contains iron. An insufficient amount of this element in the body can lead to the development of anemia (see. Iron deficiency anemia). There is a relationship between the ability to synthesize a certain amount of hemoglobin (which is due to iron stores) and erythropoiesis - in all likelihood, there is a threshold value of hemoglobin concentration, without which erythropoiesis stops.
Hemoglobin synthesis begins in erythroid precursors at the stage of formation of an erythropoietin-sensitive cell. In the fetus, and then in the early postpartum period, the child develops hemoglobin F, and then, mainly, hemoglobin A. When erythropoiesis is stressed (hemolysis, bleeding), a certain amount of hemoglobin F may appear in the blood of an adult.
Hemoglobin consists of two variants of globin chains, a and p, surrounding the heme containing iron. Depending on changes in the sequences of amino acid residues in globin chains, the chemical and physical properties of hemoglobin change, under certain conditions it can crystallize and become insoluble (for example, hemoglobin S in sickle cell anemia).
Properties of red blood cells
Red blood cells have several properties. The best known is the transport of oxygen (O₂) and carbon dioxide (CO₂). It is carried out by hemoglobin, which binds alternately with one and the other gas, depending on the voltage of the corresponding gas in the environment: in the lungs - oxygen, in the tissues - carbon dioxide. The chemistry of the reaction consists in the displacement and replacement of one gas by another from the bond with hemoglobin. In addition, erythrocytes are carriers of nitric oxide (NO), which is responsible for vascular tone and is also involved in cell signaling and many other physiological processes.
Erythrocytes have the ability to change their shape when passing through small-diameter capillaries. The cells spread out, twist into a spiral. The plasticity of erythrocytes depends on various factors, including the structure of the erythrocyte membrane, the type of hemoglobin it contains, and the cytoskeleton. In addition, the erythrocyte membrane is surrounded by a kind of "cloud" of various proteins that can change deformability. These include immune complexes, fibrinogen. These substances change the charge of the erythrocyte membrane, attach to receptors, and accelerate the sedimentation of erythrocytes in a glass capillary.
In the case of thrombus formation, erythrocytes are the centers of formation of fibrin strands, this can not only change deformability, cause their aggregation, sticking into coin columns, but also tear red blood cells into fragments, tear off pieces of membranes from them.
The erythrocyte sedimentation reaction (ESR) reflects the presence on their surface of a charge that repels erythrocytes from each other. Appearance during inflammatory reactions, with activation of coagulation, etc. a dielectric cloud around the erythrocyte leads to a decrease in repulsive forces, as a result of which the erythrocytes begin to settle faster in a vertically placed capillary. If the capillary is tilted 45 °, then the repulsive forces act only during the passage of erythrocytes across the lumen of the capillary. When the cells reach the wall, they roll down it without encountering resistance. As a result, in a tilted capillary, the erythrocyte sedimentation rate increases tenfold.
Sources:
1. Anemic syndrome in clinical practice / P.А. Vorobiev, - M., 2001;
2. Hematology: The latest reference book / Ed. K.M. Abdulkadyrov. - M., 2004.
1. Functions of blood cells. The functions of erythrocytes. Properties of erythrocytes. The Embden-Meyerhof cycle. The structure of erythrocytes.
2. Hemoglobin. Types (types) of hemoglobin. Hemoglobin synthesis. Hemoglobin function. The structure of hemoglobin.
3. Aging of erythrocytes. Destruction of red blood cells. The life span of an erythrocyte. Echinocyte. Echinocytes.
4. Iron. Iron is normal. The role of iron ions in erythropoiesis. Transferrin. The body's need for iron. Iron deficiency. OZHSS.
5. Erythropoiesis. Erythroblastic islets. Anemia. Erythrocytosis.
6. Regulation of erythropoiesis. Erythropoietin. Sex hormones and erythropoiesis.
7. Leukocytes. Leukocytosis. Leukopenia. Granulocytes. Leukocyte formula.
8. Functions of neutrophilic granulocytes (leukocytes). Defensins. Cathelicidins. Acute phase proteins. Chemotactic factors.
9. Bactericidal effect of neutrophils. Granulopoiesis. Neutrophilic granulopoiesis. Granulocytosis. Neutropenia.
10. Functions of basophils. Functions of basophilic granulocytes. Normal amount. Histamine. Heparin.
Hemoglobin. Types (types) of hemoglobin. Hemoglobin synthesis. Hemoglobin function. The structure of hemoglobin.
Hemoglobin is a hemoprotein, with a molecular weight of about 60 thousand, which stains the erythrocyte red after the binding of the O2 molecule with the iron ion (Fe ++). Have men 1 liter of blood contains 157 (140-175) g hemoglobin, at women- 138 (123-153) g. Hemoglobin molecule consists of four heme subunits associated with the protein part of the molecule - globin formed from polypeptide chains.
Heme synthesis proceeds in the mitochondria of erythroblasts. Globin chain synthesis carried out on polyribosomes and controlled by genes of the 11th and 16th chromosomes. Hemoglobin synthesis scheme in humans is shown in Fig. 7.2.
Hemoglobin containing two a- and two B-chains is called A-type (from adult - adult). 1 g of A-type hemoglobin binds 1.34 ml of O2. In the first three months of the life of a human fetus, the blood contains embryonic hemoglobins of the type Gower I (4 epsilon chains) and Gower II (chains 2a and 25). Then formed hemoglobin F(from faetus - fruit). Its globin is represented by two chains a and two B. Hemoglobin F has 20-30% higher affinity for O2 than hemoglobin A, which contributes to a better supply of oxygen to the fetus. At the birth of a child, up to 50-80% of hemoglobin is presented hemoglobin F and 15-40% - type A, and by 3 years the level hemoglobin F decreases to 2%.
Hemoglobin compound with a 02 molecule is called oxyhemoglobin. Hemoglobin affinity to oxygen and the dissociation of oxyhemoglobin (the detachment of oxygen molecules from oxyhemoglobin) depend on the oxygen tension (PO2), carbon dioxide (PCO2) in the blood, blood pH, blood temperature and the concentration of 2,3-DPG in erythrocytes. So, the affinity is increased by an increase in PO2 or a decrease in PCO2 in the blood, a violation of the formation of 2,3-DPG in erythrocytes. On the contrary, an increase in the concentration of 2,3-DPG, a decrease in blood PO2, a shift in pH to the acidic side, an increase in PCO2 and blood temperature - reduce the affinity of hemoglobin for oxygen, thereby facilitating its release to tissues. 2,3-DPG binds to β-chains hemoglobin, facilitating the detachment of 02 from the hemoglobin molecule.
An increase in the concentration of 2,3-DPG is observed in people trained for long-term physical work, adapted to a long stay in the mountains. Oxyhemoglobin that gave up oxygen is called reduced oxygen, or deoxyhemoglobin... In a state of physiological rest in humans, hemoglobin in arterial blood is 97% saturated with oxygen, in venous blood - by 70%. The more pronounced the oxygen consumption by the tissues, the lower the oxygen saturation of the venous blood. For example, with intense physical work, the oxygen consumption by the muscle tissue increases several tens of times and the oxygen saturation of the venous blood flowing from the muscles decreases to 15%. The content of hemoglobin in a single erythrocyte is 27.5-33.2 picograms. A decrease in this value indicates hypochromic(i.e., decreased), increase - o hyperchromic(i.e. increased) hemoglobin content in erythrocytes. This indicator has diagnostic value. For instance, erythrocyte hyperchromia characteristic of B | 2-deficiency anemia, hypochromia- for iron deficiency anemia.
Porphyrias - a heterogeneous group of diseases caused by impaired heme synthesis due to a deficiency of one or more enzymes.
Porphyria classifications
There is no uniform classification of porphyrias. Porphyrias are divided for reasons:
Hereditary... They arise with a defect in the gene of the enzyme involved in the synthesis of heme;
Acquired... They arise with the inhibitory effect of toxic compounds (hexochlorobenzene, heavy metal salts - lead) on the enzymes of heme synthesis.
Depending on the preferred localization of the enzyme deficiency (in the liver or erythrocytes), porphyrin is divided into:
hepatic- the most common type of porphyrin is acute intermittent porphyria (AKI), late cutaneous porphyria, hereditary coproporphyria, mosaic porphyria;
erythropoietic- congenital erythropoietic porphyria (Gunther's disease), erythropoietic protoporphyria.
Depending on the clinical picture, porphyrias are divided into:
chronic.
The negative consequences of porphyrias are associated with a deficiency of heme and the accumulation in tissues and blood of intermediate products of heme synthesis - porphyrinogens and their oxidation products. In erythropoietic porphyrias, porphyrins accumulate in normoblasts and erythrocytes, in hepatic porphyrias - in hepatocytes.
For each type of porphyria, there is a certain level of enzymatic defect, as a result, products synthesized above this level accumulate. These foods are the main diagnostic markers of the disease.
Porphyrinogens are poisonous; in severe forms of porphyrias, they cause neuropsychiatric disorders, dysfunctions of the RES and skin damage.
Neuropsychiatric disorders in porphyrias are associated with the fact that aminolevulinate and porphyrinogens are neurotoxins.
In the skin, in the sun, porphyrinogens are readily converted to porphyrins. Oxygen, when interacting with porphyrins, goes into a singlet state. Singlet oxygen stimulates lipid peroxidation of cell membranes and cell destruction; therefore, porphyrias are often accompanied by photosensitization and ulceration of open skin areas.
Porphyrinogens are neither colored nor fluorescent, while porphyrins exhibit intense red fluorescence under ultraviolet light. Excess porphyrins, which are excreted in the urine, gives it a dark color ("porphyrin" in Greek means purple).
Sometimes, with mild forms of hereditary porphyria, the disease may be asymptomatic, but taking drugs that induce the synthesis of aminolevulinate synthase can exacerbate the disease. In some cases, symptoms of the disease do not appear until puberty, when an increase in the formation of β-steroids induces the synthesis of aminolevulinate synthase. Porphyrias are also observed in case of poisoning with lead salts, since lead inhibits aminolevulinate dehydratase and ferrochelatase. Some halogenated herbicides and insecticides are inducers of the synthesis of aminolevulinate synthase, so their ingestion is accompanied by symptoms of porphyria.
Types of porphyrias
Acute intermittent porphyria (AKI) - cause - a defect in the gene encoding PBG - deaminase. It is inherited in an autosomal dominant manner. There is an accumulation of early precursors of heme synthesis: 5-ALA (5-ALA) and porphobilinogen (PBG).
Colorless PBG in the light turns into porfibilin and porphyrin, they make the urine dark. ALA has a neurotoxic effect, leading to flaccid paralysis of the limbs and paresis of the respiratory muscles. The latter causes acute respiratory failure. The disease manifests itself in middle age, is provoked by the intake of analgesics, sulfa drugs, as they increase the synthesis of ALA - synthase.
Clinical symptoms are acute abdominal pain, vomiting, constipation, cardiovascular disorders, neuropsychiatric disorders. There is no hypersensitivity to light, since the metabolic disorder occurs at the stage preceding the formation of uroporphyrinogen.
For treatment, the drug normosang is used - heme arginate. The action is based on the fact that heme, through the mechanism of negative feedback, blocks the translation of ALA - synthase, and, consequently, the synthesis of ALA and PBG decreases, which is how the relief of symptoms is achieved.
Congenital erythropoietic porphyria is an even rarer congenital disorder inherited in an autosomal recessive manner. The molecular nature of this disease is not exactly known; it was found, however, that it is characterized by a certain imbalance in the relative activities of uroporphyrinogen-III-cosynthase and uroporphyrinogen-1-synthase. The formation of uroporphyrinogen I quantitatively significantly exceeds the synthesis of uroporphyrinogen III-normal isomer on the heme synthesis pathway. Although the genetic disorder extends to all cells, it manifests itself, for unknown reasons, predominantly in erythropoietic tissue. Patients with congenital erythropoietic porphyria excrete large amounts type isomersI uroporphyrinogen and coproporphyrinogen; in urine, both of these compounds spontaneously oxidize to uroporphyrin I and coproporphyrin I-red fluorescent pigments. It was reported that there was a slight increase in the concentration of uroporphyrin III, but the ratio of type I and III isomers was approximately 100: 1. Circulating red blood cells contain a large amount of uroporphyrin 1, however, the highest concentration of this porphyrin was noted in bone marrow cells (but not in hepatocytes).
It is noted photosensitivity of the skin, due to the nature of the absorption spectrum of porphyrin compounds, which are formed in large quantities. In patients, cracks in the skin are noted, hemolytic phenomena are often observed.
Hereditary coproporphyria -Autosomal dominant disorder due to deficiency coproporphic genoxidase- a mitochondrial enzyme responsible for the conversion of coproporphyrinogen III into protoporphyrinogen IX. Coproporphyrinogen III is excreted in large quantities from the body in feces, and due to its solubility in water, it is excreted in large quantities in the urine. Like uroporphyrinogen, coproporphyrinogen is rapidly oxidized in light and air, turning into the red pigment coproporphyrin.
The limited ability of this disease to synthesize heme (especially under stressful conditions) leads to derepression of ALA-siitase. As a result, there is an excessive formation of ALA and porphobilinogen, as well as other intermediates on the pathway of synthesis of the topic, formed at the stages preceding the hereditarily blocked stage. Accordingly, in patients with hereditary coproporphyria, all the signs and symptoms associated with an excess of ALA and porphobilinogen, which are characteristic of intermittent acute porphyria, are found, but in addition they have increased photosensitivity due to the presence of excess amounts of coproporphyrinogens and uroporphyrinogens. In this disease, the administration of hematin can also cause at least partial repression of ALA synthase and amelioration of symptoms caused by the overproduction of heme biosynthetic intermediates.
Mosaic porphyria , or hereditary photoporphyria, is an autosomal dominant disorder in which there is a partial blockage of the enzymatic conversion of protoporphyrinogen to heme. Normally, this transformation is carried out by two enzymes, protoporphyrinogen oxidase and ferrochelatase, localized in mitochondria. Judging by the data obtained on the culture of skin fibroblasts, in patients with porphyria mosaic, the content of protoporphyrinogen oxidase is only half the normal amount. In patients with porphyria mosaic, there is a relative deficiency in the heme content under stressful conditions, as well as a derepressed state of the hepatic ALA synthase. As noted above, the increased activity of ALA synthase leads to the overproduction of all heme synthesis intermediates at the sites before the blocked stage. Thus, patients with porphyria mosaic excrete excessive amounts of ALA, porphobilinogen, uroporphyrin and coproporphyrin in the urine, and uroporphyrin, coproporphyrin and protoporphyrin in the feces. The urine of patients is pigmented and fluorescent, and the skin is sensitive to light in the same way as in patients with tardive cutaneous porphyria (see below).
Late cutaneous porphyria , is probably the most common form of porphyria. Usually it is associated with some kind of liver damage, especially with excessive alcohol consumption or overload with iron ions. The nature of the metabolic disorder has not been precisely established, but the probable cause is a partial deficiency of uroporphyrinogen decarboxylase. The disorder appears to be transmitted as an autosomal dominant trait, but genetic penetrance is variable and in most cases depends on the presence of liver dysfunctions. According to the predictions, urine contains increased amounts of uroporphyrins of type I and III; at the same time, urinary excretion of ALA and porphobilinogen is relatively rare. Sometimes the urine contains a very significant amount of porphyrins, giving it a pinkish tint; when acidified, it most often gives pink fluorescence in the ultraviolet region.
The liver contains large amounts of porphyrins and therefore fluoresces strongly, while erythrocytes and bone marrow cells do not have fluorescence. The main clinical manifestation in tardive cutaneous porphyria is increased photosensitivity of the skin. In patients, there is neither increased activity of ALA synthase, nor, accordingly, excess content in the urine of porphobilinogen and ALA; this correlates with the absence of acute attacks characteristic of acute intermittent porphyria.
Protoporphyria , or erythropoietic protoporphyria appears to be due to dominantly inherited deficiency ferrochelatase in the mitochondria of all tissues; clinically, this disease manifests itself as acute urticaria caused by exposure to sunlight. Erythrocytes, plasma, and feces contain elevated amounts of protoporphyrin IX, and reticulocytes (immature red blood cells) and skin (when examined by biopsy) often fluoresce with red light. The liver probably also contributes to the increase in the formation of protoporphyrin IX, but urinary excretion of porphyrins and their precursors is not observed.
Hemoglobin synthesis
The heme synthesized in mitochondria is induced by the synthesis of globin chains on polyribosomes. Globin chain genes are located on chromosomes 11 and 16.
Globin chains form globules and bind to heme. 4 globules are non-covalently combined to form hemoglobin.
Hemoglobin begins to be synthesized at the stage of basophilic erythroblast, and ends in reticulocytes. In reticulocytes, the synthesis of purines, pyrimidines, phosphatides, lipid also takes place. A sensitive biochemical indicator for distinguishing reticulocytes from mature cells is the loss of glutaminase by the latter. Glutamine in reticulocytes is a carbon source for porphyrin synthesis and nitrogen for purine synthesis.
Hemoglobin structure
Hemoglobin - tetrameric chromoprotein, has a mass of 64500 Da, consists of 4 hemes and 4 globins. Globins are represented by polypeptide chains of various types,,, , etc.-chain contains 141 AA, and-chain - 146 AA. Separate sections of polypeptide chains form right-handed-helices, a special arrangement in the space of which forms globules. Globule -subunits contain 8-helices, a-subunits –7. Heme is located in the gaps between the E and F coils of globin, attaching through histidine F 8 to the F helix by means of the 5 coordination bond of iron. The hydrophobic amino acid residues surrounding the heme prevent the oxidation of iron by water. 4 globules with the participation of hydrophobic, ionic and hydrogen bonds form a spherical hemoglobin tetramer. The strongest bonds, mainly due to hydrophobic bonds, are formed between-and-globules. As a result, 2 dimers 1 1 and 2 2 are formed. Dimers are connected to each other mainly by polar (ionic and hydrogen) bonds, therefore the interaction of dimers depends on pH. Dimers easily move relative to each other. In the center of the tetramer, the globules are loosely adjacent to each other, forming a cavity.
Hemoglobin functions
Provide oxygen transfer from the lungs to the tissues. About 600 liters per day;
Participates in the transfer of carbon dioxide and protons from tissues to the lungs;
Regulates blood count.
Any pathology in iron metabolism is accompanied by the development anemia – a painful condition characterized by either a decrease in the number of red blood cells or a decrease in the contribution of hemoglobin. This is one of the most common ailments. It occurs as a result of a variety of reasons:
a) due to a lack of iron in the diet (in vegetarians, during fasting, when using different diets for weight loss, in pregnant women, during breastfeeding, in rapidly growing adolescents);
b) due to impaired absorption in the gastrointestinal tract (with hyposecretion of hydrochloric acid, proteases, after subtotal gastrectomy, with shifts in the balance of nutrients - a lack of ascorbate, succinate, an excess of phytic acid, fiber, with damage to the intestinal mucosa in peptic ulcer disease, diaphragmatic hernia , ulcerative colitis, after treatment with salicylates, steroids, with helminthiasis, especially with lesions with whipworm, hookworm);
c) due to insufficient iron reserves;
d) due to changes in individual links of the metabolism of this transition metal (with inhibition of the activity of heme synthesis enzymes);
e) after an increased release of ions of this metal from the body (with acute and chronic blood loss, after heavy menstruation - polymenorrhea, with hemorrhoids, various ulcers in the stomach, intestines, after repeated episodes of hemoptysis).
The main clinical signs of anemia: weakness, palpitations, fatigue, absent-mindedness, pallor, shortness of breath.
Depending on the degree of preservation of the amount of iron in the body, Fe-deficient, Fe-sufficient, Fe-excess anemias are secreted. About 98 - 99% of all cases of such diseases occur in the first option. Others are based on abnormalities in the use of iron in heme synthesis. The transition metal ions not included in this compound begin to be deposited in the form hemosiderin (hereditary and acquired hemochromatosis ) in organs and tissues (liver, pancreas, myocardium, joints, skin) with the subsequent suppression of their functions. The following triad of signs can be traced: cirrhosis of the liver, diabetes mellitus, bronze coloration of the skin (bronze diabetes). And since symptoms of anemia develop in parallel (due to a deficiency of hemoglobin heme in erythrocytes), it is especially dangerous to use iron preparations as therapeutic agents.
An example of such anemias is methylmalonic aciduria , which is based on genetic damage to the work of the B12-containing enzyme - methylmalonyl-CoA mutase , responsible for the isomerization of methylmalonyl-Co A to succinyl-CoA - one of the substrates in the genesis of heme.
Pathogenesis Heilmeyer's disease comes down to the fact that the function of the gene responsible for the synthesis of transferrin is blocked. In its absence, the system of iron release in the bone marrow does not work, in this regard, the formation of heme is suppressed, anemia develops. The disease is inherited in an autosomal recessive manner.
Diseases based on damage in heme synthesis are named porphyrias ... Depending on the localization of disorders, erythropoietic (damage in the metabolism of porphyrins in the bone marrow) and hepatic (similar changes in hepatocytes) types are distinguished. Most often this is due to genetics, less often it is acquired in nature. Currently, blocks of all enzymes participating in heme synthesis have been registered.
Porphyria (more precisely, its hereditary erythropoietic form) was first described by Schultz (1874) and Gunther (1911). However, the historical chronicles of the Middle Ages have preserved descriptions of families whose members showed features characteristic of severe forms of this suffering, manifested by skin, neurological and abdominal symptoms (acute abdominal symptoms, epileptic seizures, polyneuritis, hallucinations, blindness), as well as an abnormally high release of porphyrins with urine or feces. Some signs of the disease are a red tint of teeth and bones, a peculiar color of the skin, altered by blisters, ulcers and scars; the nocturnal lifestyle caused by photodermatitis, the spontaneous glow of some tissues and secretions of the patient, the whims of taste associated with anemia are so vivid and unusual that they recall descriptions of the appearance and behavior of mythical ghouls or vampires.
More rarely, acquired porphyrias are found, the causes of which are poisoning with salts of heavy metals, which, interacting with the sulfhydryl groups of aminolevulinate synthase or ferrochelatase, suppress their activity in the genesis of heme. As a result, protoporphyrin accumulates in erythrocytes, iron content increases in blood plasma, it is deposited in organs and tissues, provoking the formation of hemosiderosis.
Naturally, the pathology of globin synthesis is hereditary. There are two main forms of disorders: single amino acid damage in the protein structure ( hemoglobinosis ) and suppression of the production of any polypeptide chain of globin ( thalassemias ).
Currently, more than 300 types of pathological hemoglobins have been described. The first modified hemoproteins were named using Latin letters (C, D, E, M, S), but when the number of modified Hb exceeded the number of letters in the alphabet, they began to be named after the place of discovery (Kansas, Boston, San Yose, Hiroshima, Richmond, etc. ).
The heterogeneous nature of genetic damage (replacement, insertion, frame shift, chain elongation, etc.) leads to various consequences (the affinity for oxygen changes, the stability of erythrocytes decreases, which is manifested by increased hemolysis, cyanosis). This is determined not only by the severity of shifts in the nucleotide sequence, but also by the nature of the changed amino acids. If analogs are replaced, for example, glutamate for aspartate, then this option does not manifest itself in any way. The carriers of Hb San Jose are also practically healthy, in which in the 7th position of the beta chain glutamate is replaced by glycine, which only affects the electrophoretic mobility of this protein.
If the structure of the mutated site changes abruptly, then the likelihood of developing serious clinical signs is high. Particularly dangerous are lesions located in the contact areas (the places where individual subunits bind into tetramers) or in the pockets where the heme is located. In these cases, the complexation of the heterogeneous tetramer is disrupted, which affects the viability of the embryo and increases the likelihood of miscarriage. An example is Hb Philly, in which phenylalanine replaces tyrosine, which in a normal protein forms a hydrogen bond with other subunits. After such a mutation, the existence of a genetically altered tetramer becomes impossible - the micelle falls apart. Boston Hb is characterized by the substitution of tyrosine for histidine at position 58 of the alpha chain. Histidine, on the other hand, usually forms a coordination bond with heme iron, and tyrosine oxidizes the ion to iron phenolate; the resulting methemoglobin provokes hypoxia.
In Genowa hemoglobin, due to the loss of valine, the fragment is displaced, the remainder of the glutamate is inside the micelle, the site is deformed, which reduces the affinity for oxygen, and cyanosis develops.
More favorable consequences are possible if mutations affect the amino acids that create the micelle surface. A classic example is Hb S, in the 6th position of the beta chain of which glutamate is replaced by valine, that is, an acidic compound is replaced by a hydrophobe. And since the damage is on the surface, such a change reduces the charge and solubility of hemoglobin, and its individual molecules, colliding, stick together due to the formation of hydrophobic interactions of valines of different micelles. Such aggregation lengthens the hemoprotein filaments, which further reduces the solubility. This phenomenon affects the shape of red blood cells: they take the shape of a sickle (Sickle cell - sickle cell). Hence the disease is called sickle-shaped anemia, or hemoglobinosis S . The damaged erythrocyte membrane is less stable, which provokes hemolysis, thrombosis. Patients are characterized by hemolytic crises with acute pain syndrome, symptoms of liver damage, intense jaundice, and probably the formation of stones in the biliary tract. The frequency of the presence of such a hemoprotein in the United States is 8-9% (in African Americans), and in some regions of Greece it reaches 40%.
Thalassemias(thalassa - sea) - hereditary diseases, which, as noted above, are based on the block of synthesis of the whole globin chain. In this case, micelles are synthesized in erythroid cells, often representing homogeneous tetramers, for example, consisting only of beta or gamma chains. Hemoglobin H, consisting of 4 beta subunits, is only able to bind oxygen, but not to give. With the inhibition of the synthesis of alpha chains, the homozygous embryo is not viable, a miscarriage occurs in early pregnancy. Child with beta thalassemia (Cooly disease) (a block of beta chain genesis) is born practically healthy (after all, hemoglobin, including alpha and gamma chains, is needed for the growth and development of the fetus). Every month, pathological signs begin to appear, determined by the degree of hypoxia, increased lipid peroxidation, damage to erythrocyte membranes (severe hemolytic anemia, persistence of a high HbF content (up to 20-30%), impaired physical and mental development, the child refuses to breast, the face acquires Mongoloid features - the cheekbones protrude forward, the base of the bridge of the nose is pressed in, the nose becomes flattened). For an older age, hemolytic crises, a febrile state are characteristic, and heart failure may develop.
The spread of these diseases varies by region. They are especially common on the shores of the seas (Mediterranean, Black). In Transcaucasia, up to 10% of carriers of such genes are registered.
DISHEMOGLOBINEMIA
Pathological conditions are relatively common, which are based on imbalances in various forms of hemoglobin. In the erythrocytes of a healthy adult, the level methemoglobin does not exceed 2% of the total content. However, under the influence of various nitrogen oxides, inorganic nitrates, organic nitro compounds (amyl nitrite, nitrobenzene, nitrophenol, trinitrotoluene, nitroaniline), amino derivatives (hydroxylamine, phenylhydrazine, aminophenols, p-aminobenzoic acid, aniline), chlorates, chromates, permanganates, pyrone some drugs (nitroglycerin, anestezin, furadonin, barbiturates, aspirin, etc.), paints with an oxidizing ability - its concentration increases sharply ( met-hemoglobinemia ). As a result, the main function of hemoglobin is disrupted - the transfer of oxygen from the lungs to the tissues is blocked and hypoxia develops.
All methemoglobin-formers, reducing the osmotic resistance of erythrocytes, accelerate their hemolysis. During the oxidation of Hb to methemoglobin, active oxygen radicals are generated, which can take part in the processes of oxidative damage to erythrocytes. LPO is activated, lipid metabolism of red blood cell membranes is disturbed, the balance in the LPO - AOD system is shifted. The main symptom is cyanosis; if the content of methemoglobin exceeds 30%, weakness, dizziness, tachycardia, headaches occur; when it accumulates up to 50%, cardiovascular failure develops. The risk of nitrite poisoning increases with the use of vegetables, sausages, stewed meat, and poor-quality drinking water. Cases of hereditary methemoglobinemias (in carriers of Hb Boston) have also been described (see above).
The group of blood poisons that form pathological pigments includes carbon monoxide (carbon monoxide). Penetrating into the body, CO is absorbed by erythrocytes, interacts with the hemoglobin iron, forming a fairly stable compound - carboxyhemoglobin , the value of which in nonsmokers does not exceed 0.25% of the total amount of the basic protein of erythrocytes. In the blood of smokers, its numbers increase to 6-7%. At a higher partial pressure of carbon monoxide ( carboxyhemoglobinemia ) oxygenation of hemoprotein is inhibited, hypoxia develops. In addition, CO has the ability to come into contact with other heme-containing proteins (myoglobin, cytochromes, peroxidase, catalase), disrupting their functions.