Hidroliza. Hidroliza e estereve Esteret dhe hidroliza e tyre
Esteret janë derivate të acideve në të cilat hidrogjeni acidik zëvendësohet nga radikale alkil (ose përgjithësisht hidrokarbur).
Esteret ndahen në varësi të cilit acid rrjedhin (inorganik ose karboksilik).
Në mesin e estereve, një vend të veçantë zënë esteret natyrore - yndyrat dhe vajrat, të cilat formohen nga glicerina e alkoolit trihidrik dhe acidet yndyrore më të larta që përmbajnë një numër çift atomesh karboni. Yndyrnat janë pjesë e organizmave bimorë dhe shtazorë dhe shërbejnë si një nga burimet e energjisë së organizmave të gjallë, i cili lirohet gjatë oksidimit të yndyrave.
Formula e përgjithshme për esteret e acidit karboksilik është:
ku R dhe R" janë radikale hidrokarbure (në esteret e acidit formik, R është një atom hidrogjeni).
Formula e përgjithshme për yndyrnat:

ku R", R", R"" janë radikale karboni.
Yndyrnat janë "të thjeshta" dhe "të përziera". Përbërja e yndyrave të thjeshta përfshin mbetje të të njëjtave acide (d.m.th. R’ = R "= R""), përbërja e yndyrave të përziera përfshin të ndryshme.
Acidet yndyrore më të zakonshme që gjenden në yndyrna janë:
1. Acidi butirik CH 3 - (CH 2) 2 - COOH
3. Acidi palmitik CH 3 - (CH 2) 14 - COOH
4. Acidi stearik CH 3 - (CH 2) 16 - COOH
5. Acidi oleik C 17 H 33 COOH
CH 3 -(CH 2) 7 -CH === CH-(CH 2) 7 -COOH
6. Acidi linoleik C 17 H 31 COOH
CH 3 -(CH 2) 4 -CH \u003d CH-CH 2 -CH \u003d CH-COOH
7. Acidi linolenik C 17 H 29 COOH
CH 3 CH 2 CH \u003d CHCH 2 CH \u003d\u003d CHCH 2 CH \u003d CH (CH 2) 4 COOH
Esteret karakterizohen nga llojet e mëposhtme të izomerizmit:
1. Izomerizmi i zinxhirit të karbonit fillon në mbetjen e acidit me acid butanoik, në mbetjen e alkoolit - me alkoolin propil, për shembull, izobutirati etil, acetati propil dhe acetati izopropil janë izomerë të butiratit të etilit.
2. Izomerizmi i pozicionit të grupit ester -CO-O-. Ky lloj izomerizmi fillon me estere që përmbajnë të paktën 4 atome karboni, si acetati etil dhe metil propionati.
3. Izomerizmi ndërklasor, për shembull acidi propanoik është izomerik ndaj metil acetatit.
Për esteret që përmbajnë acid të pangopur ose alkool të pangopur, dy lloje të tjera të izomerizmit janë të mundshme: izomerizmi i pozicionit të lidhjes së shumëfishtë dhe cis-, trans-izomeria.
Esteret e acideve më të ulëta karboksilike dhe alkooleve janë lëngje të paqëndrueshme, të patretshme në ujë. Shumë prej tyre kanë një erë të këndshme. Kështu, për shembull, butil butirati ka erë ananasi, izoamil acetati ka erë dardhe, etj.
Esteret e acideve yndyrore më të larta dhe alkooleve janë substanca dylli, pa erë, të patretshme në ujë.
Aroma e këndshme e luleve, frutave, manave është kryesisht për shkak të pranisë së estereve të caktuara në to.
Yndyrnat janë të shpërndara gjerësisht në natyrë. Së bashku me karbohidratet dhe proteinat, ato janë pjesë e të gjithë organizmave bimorë dhe shtazorë dhe përbëjnë një nga pjesët kryesore të ushqimit tonë.
Sipas gjendjes së tyre të grumbullimit në temperaturën e dhomës, yndyrat ndahen në të lëngshme dhe të ngurta. Yndyrnat e ngurta, si rregull, formohen nga acide të ngopura, yndyrat e lëngshme (ato shpesh quhen vajra) janë të pangopura. Yndyrnat janë të tretshme në tretës organikë dhe të patretshëm në ujë.
1. Reaksioni i hidrolizës, ose saponifikimit. Meqenëse reaksioni i esterifikimit është i kthyeshëm, prandaj, në prani të acideve, reagimi i kundërt i hidrolizës vazhdon:
Reaksioni i hidrolizës katalizohet gjithashtu nga alkalet; në këtë rast, hidroliza është e pakthyeshme, pasi acidi që rezulton me alkali formon një kripë:
2. Reaksioni i shtimit. Esteret që përmbajnë një acid të pangopur ose alkool në përbërjen e tyre janë të afta për reaksione shtesë.
3. Reagimi i rikuperimit. Reduktimi i estereve me hidrogjen çon në formimin e dy alkooleve:
4. Reaksioni i formimit të amideve. Nën veprimin e amoniakut, esteret shndërrohen në amide acide dhe alkoole:
Faturë. 1. Reagimi i esterifikimit:
Alkoolet reagojnë me acidet minerale dhe organike për të formuar estere. Reaksioni është i kthyeshëm (procesi i kundërt është hidroliza e estereve).

Reaktiviteti i alkooleve monohidrike në këto reaksione zvogëlohet nga primar në terciar.
2. Ndërveprimi i anhidriteve të acidit me alkoolet:

3. Ndërveprimi i halogjeneve acide me alkoolet:


Mekanizmi i hidrolizës:

Yndyrnat e lëngëta shndërrohen në të ngurta nga një reaksion hidrogjenizimi. Hidrogjeni shtohet në vendin e thyerjes së lidhjes së dyfishtë në radikalet hidrokarbure të molekulave të yndyrës:

Reagimi vazhdon kur nxehet nën presion dhe në prani të një katalizatori - nikel i ndarë imët. Produkti i hidrogjenizimit është yndyra e ngurtë (dhjamë derri artificiale), e quajtur dhjamë, e cila përdoret për të prodhuar sapun, stearinë dhe glicerinë. Margarina - yndyrë e ngrënshme, përbëhet nga një përzierje e vajrave të hidrogjenizuar (luledielli, fara pambuku, etj.), yndyrat shtazore, qumështi dhe disa substanca të tjera (kripë, sheqer, vitamina, etj.).
Një veti e rëndësishme kimike e yndyrave, si të gjithë esteret, është aftësia për t'iu nënshtruar hidrolizës (saponifikimit). Hidroliza vazhdon lehtësisht kur nxehet në prani të katalizatorëve - acideve, alkaleve, oksideve të magnezit, kalciumit, zinkut:

Reagimi i hidrolizës së yndyrave është i kthyeshëm. Sidoqoftë, me pjesëmarrjen e alkaleve, pothuajse i vjen fundi - alkalet shndërrojnë acidet që rezultojnë në kripëra dhe në këtë mënyrë eliminojnë mundësinë e ndërveprimit të acideve me glicerinë (reagim i kundërt).
| " |
Esteret të quajtura derivate funksionale të acideve karboksilike të formulës së përgjithshme RC(O)OR" .
Esteret e acideve karboksilike (si dhe acidet sulfonike) emërtohen në mënyrë të ngjashme me kripërat, vetëm se në vend të emrit të kationit përdoret emri i alkilit ose arilit përkatës, i cili vendoset para emrit të anionit dhe shkruhet së bashku. me të. Prania e grupit të esterit -COOR mund të shprehet gjithashtu në një mënyrë përshkruese, për shembull, "R-ester i acidit (të tillë dhe të tillë)" (kjo metodë është më pak e preferuar për shkak të rëndimit të saj):
Esteret e alkooleve më të ulëta dhe acideve karboksilike janë lëngje të paqëndrueshme, me erë të këndshme, të dobëta të tretshme në ujë dhe mirë - në shumicën e tretësve organikë. Erërat e estereve të kujtojnë aromat e frutave të ndryshme, prandaj në industrinë ushqimore prej tyre përgatiten esenca që imitojnë aromat e frutave. Rritja e paqëndrueshmërisë së estereve përdoret për qëllime analitike.
Hidroliza. Më e rëndësishmja nga reaksionet e acilimit është hidroliza e estereve me formimin e një alkooli dhe një acidi karboksilik:
Reaksioni kryhet në mjedise acidike dhe alkaline. acid i katalizuar hidroliza e esterit - reagimi i kundërt i esterifikimit, vazhdon sipas të njëjtit mekanizëm A AC 2:
Nukleofili në këtë reaksion është uji. Zhvendosja e ekuilibrit drejt formimit të alkoolit dhe acidit sigurohet nga shtimi i ujit të tepërt.
Hidroliza alkaline është e pakthyeshme; gjatë reaksionit, një mol alkali konsumohet për mol eter, d.m.th., alkali në këtë reaksion vepron si një reagent i konsumueshëm dhe jo një katalizator:
Hidroliza e estereve në mjedis alkalik vazhdon nëpërmjet mekanizmit bimolekular acil B AC 2 nëpërmjet formimit të një ndërmjetësi tetraedral (I). Pakthyeshmëria e hidrolizës alkaline sigurohet nga ndërveprimi praktikisht i pakthyeshëm acid-bazë i acidit karboksilik (II) dhe jonit alkoksid (III). Anioni që rezulton i acidit karboksilik (IV) është në vetvete një nukleofil mjaft i fortë dhe për këtë arsye nuk i nënshtrohet sulmit nukleofilik.
Amonoliza e estereve. Amidet fitohen me amonolizë të estereve. Për shembull, nën veprimin e amoniakut ujor në dietil fumarat, formohet amidi i plotë i acidit fumarik:
Në amonolizën e estereve me aminat me nukleofilicitet të ulët, këto të fundit shndërrohen fillimisht në amide të metaleve alkali ose tokësore alkaline:

Amidet e acideve karboksilike: nomenklatura; struktura e grupit amide; vetitë acid-bazike; hidroliza acide dhe alkaline; ndarja nga hipobromitet dhe acidi azotik; dehidratim ndaj nitrileve; identifikimi kimik.
Amidet të quajtura derivate funksionale të acideve karboksilike të formulës së përgjithshme R-C (O) -NH 2- n R "n, ku n = 0-2. Në amidet e pazëvendësuara, mbetja e acilit lidhet me një grup amino të pazëvendësuar, në amidet e zëvendësuara me N, një nga atomet e hidrogjenit zëvendësohet nga një radikal alkil ose aril, në N, N-zëvendësuar - me dy.
Përbërjet që përmbajnë një, dy ose tre grupe acil të lidhura me atomin e azotit quhen përgjithësisht amide (përkatësisht parësore, dytësore dhe terciare). Emrat e amideve primare me një grup të pazëvendësuar - NH 2 rrjedhin nga emrat e radikalëve përkatës acil duke zëvendësuar prapashtesën -vaj (ose -yl) me -amid. Amidet e formuara nga acidet me prapashtesën -acid karboksilik marrin prapashtesën -karboksamide. Amidet e acideve sulfonike emërtohen gjithashtu sipas acideve të tyre përkatëse, duke përdorur prapashtesën -sulfonamide.
Emrat e radikalëve RCO-NH- (si dhe RSO 2 -NH-) janë formuar nga emrat e amideve, duke ndryshuar prapashtesën -amide në -amido-. Ato përdoren nëse ka një grup më të vjetër në pjesën tjetër të molekulës ose zëvendësimi ndodh në një strukturë më komplekse se radikali R:

Në emrat e amideve primare N-zëvendësuese RCO-NHR" dhe RCO-NR"R" (si dhe sulfonamide të ngjashme), emrat e radikalëve R" dhe R" tregohen përpara emrit të amidit me simbolin N. -:
Amidet e këtij lloji shpesh quhen amide sekondare dhe terciare, gjë që nuk rekomandohet nga IUPAC.
Amideve të zëvendësuara me N-fenil u jepet prapashtesa -anilid në emrat e tyre. Pozicioni i zëvendësuesve në mbetjen e anilinës tregohet me numra me goditje:
Përveç kësaj, janë ruajtur emra gjysëm sistematikë në të cilët prapashtesa -amide lidhet me bazën e emrit latin të acidit karboksilik (formamid, acetamid), si dhe disa i parëndësishëm emra të tillë si "anilide" (anilina të aciluara) ose "toluidide" (toluidina të aciluara).
Amidet janë substanca kristalore me pika shkrirjeje relativisht të larta dhe të dallueshme, gjë që lejon që disa prej tyre të përdoren si derivate për identifikimin e acideve karboksilike. Në raste të rralla, ato janë lëngje, për shembull, amide të acidit formik - formamide dhe N,N-dimetilformamide - tretës të njohur dipolarë aprotikë. Amidet e ulëta janë shumë të tretshme në ujë.
Amidet janë një nga më rezistentët ndaj hidrolizës derivatet funksionale të acideve karboksilike, për shkak të të cilave ato shpërndahen gjerësisht në natyrë. Shumë amide përdoren si ilaçe. Për rreth një shekull, paracetamoli dhe fenacetina, të cilat janë amide të zëvendësuara të acidit acetik, janë përdorur në praktikën mjekësore.
Struktura e amideve. Struktura elektronike e grupit amide është kryesisht e ngjashme me strukturën e grupit karboksil. Grupi amid është një sistem i konjuguar p,π në të cilin çifti i vetëm i elektroneve të atomit të azotit është i konjuguar me elektronet e lidhjes C=O π. Delokalizimi i densitetit të elektroneve në grupin e amidit mund të përfaqësohet nga dy struktura rezonancë:
Për shkak të konjugimit, lidhja C-N në amide ka pjesërisht të lidhura dyfish karakteri, gjatësia e saj është dukshëm më e vogël se gjatësia e një lidhjeje tek aminet, ndërsa lidhja C=O është disi më e gjatë se lidhja C=O në aldehide dhe ketone. Grupi amide për shkak të konjugimit ka një dizajn të sheshtë . Më poshtë janë parametrat gjeometrikë të molekulës së amidit të zëvendësuar me N, të përcaktuara duke përdorur analizën e difraksionit me rreze X:

Një pasojë e rëndësishme e natyrës pjesërisht të lidhur dyfish të lidhjes C-N është një pengesë mjaft e lartë e energjisë për rrotullimin rreth kësaj lidhjeje, për shembull, për dimetilformamidin është 88 kJ/mol. Për këtë arsye, amidet që kanë zëvendësues të ndryshëm në atomin e azotit mund të ekzistojnë si π-diastereomere. Amidet e zëvendësuara me N ekzistojnë kryesisht si Z-izomerë:

Në rastin e amideve N,N të dyzëvendësuara, raporti i izomerëve E- dhe Z varet nga vëllimi i radikalëve të lidhur me atomin e azotit. Stereoizomerët e amideve janë të paqëndrueshëm konfiguracioni, ekzistenca e tyre është vërtetuar kryesisht me metoda fiziko-kimike dhe ato janë izoluar individualisht vetëm në disa raste. Kjo për faktin se barriera e rrotullimit për amidet nuk është ende aq e lartë sa për alkenet, për të cilat është 165 kJ/mol.
Vetitë acido-bazike. Amidet kanë veti të dobëta si acidike ashtu edhe ato bazike . Baza e amideve qëndron brenda intervalit Pk BH + nga -0.3 në -3.5. Arsyeja për bazueshmërinë e reduktuar të grupit amino në amide është konjugimi i çiftit të vetëm të elektroneve të atomit të azotit me grupin karbonil. Kur ndërveprojnë me acide të forta, amidet protonohen në atomin e oksigjenit si në tretësirat e holluara ashtu edhe në ato të koncentruara të acidit. Ky lloj ndërveprimi qëndron në themel kataliza acide në reaksionet e hidrolizës amide:
Amidet e pazëvendësuara dhe N-zëvendësuara shfaqin veti të dobëta të acidit NH , të krahasueshme me aciditetin e alkooleve dhe hiqni një proton vetëm në reaksione me baza të forta.
Ndërveprimi acid-bazë qëndron në themel të formimit të amideve bashkëpunëtorët ndërmolekularë , ekzistenca e të cilave shpjegon pikat e larta të shkrirjes dhe vlimit të amideve. Ekzistenca e dy llojeve të asociacioneve është e mundur: polimere lineare dhe dimere ciklike. Mbizotërimi i një lloji ose një tjetër përcaktohet nga struktura e amidit. Për shembull, N-metilacetamidi, për të cilin preferohet konfigurimi Z, formon një lidhje lineare dhe laktamet, të cilat kanë një konfigurim E të fiksuar fort, formojnë dimerë:

Amidet N, N-Zëvendësuar formojnë dimerë për shkak të ndërveprimit dipol-dipol të 2 molekulave polare:

Reaksionet e acilimit. Për shkak të pranisë së një grupi të fortë amino dhurues të elektroneve në sistemin e amidit të konjuguar, elektrofiliteti i atomit të karbonit karbonil, dhe si rrjedhim, reaktiviteti i amideve në reaksionet e acilimit, është shumë i ulët. Aftësia e ulët aciluese e amideve shpjegohet edhe me faktin se joni amid NH 2 - është një grup largues i keq. Nga reaksionet e acilimit, hidroliza e amideve është e rëndësishme, e cila mund të kryhet në mjedise acidike dhe alkaline. Amidet janë shumë më të vështira për t'u hidrolizuar sesa derivatet e tjerë funksionalë të acideve karboksilike. Hidroliza e amideve kryhet në kushte më të rënda në krahasim me hidrolizën e estereve.
Hidroliza e acidit amide - të pakthyeshme Reaksioni që çon në formimin e një acidi karboksilik dhe një kripë amoniumi:
Në shumicën e rasteve, hidroliza acide e amideve vazhdon sipas mekanizmit Acilimi i acidit bimolekular A AC 2 , pra i ngjashëm me mekanizmin e hidrolizës acide të estereve. Pakthyeshmëria e reaksionit është për shkak të faktit se amoniaku ose amina në një mjedis acid shndërrohet në një jon amoniumi që nuk ka veti nukleofile:

Hidroliza alkaline Njësoj të pakthyeshme reagimi; si rezultat i tij, formohet një kripë e një acidi karboksilik dhe amoniak ose një aminë:
Hidroliza alkaline e amideve, si hidroliza e estereve, zhvillohet nëpërmjet mekanizmi tetraedral NË AC 2 . Reaksioni fillon me shtimin e një joni hidroksid (nukleofili) në atomin elektrofilik të karbonit të grupit amid. Anioni që rezulton (I) protonohet në atomin e azotit, dhe më pas një grup largues i mirë, një molekulë amoniaku ose amine, formohet në jonin bipolar (II). Besohet se faza e ngadaltë është prishja e ndërmjetësit tetraedral (II).
Për anilidet dhe amide të tjera me zëvendësues elektron-tërheqës në atomin e azotit, dekompozimi i ndërmjetësit tetraedral (I) mund të vazhdojë përmes formimit të dianionit (II):

Ndarja me acid azoti. Kur ndërveprojnë me acidin azotik dhe agjentët e tjerë nitrozues, amidet shndërrohen në acidet karboksilike përkatëse me rendimente deri në 90%:
Dehidratim. Amidet e pazëvendësuara nën veprimin e oksidit të fosforit (V) dhe disa reagentëve të tjerë (POC1 3, PC1 5, SOCl 2) shndërrohen në nitrile:
![]()
47. Acidet karboksilike: halogjenimi sipas Gell-Volhard-Zelinsky, duke përdorur reaksionin për sintezë a -hidroksi dhe a -aminoacidet.
Halogjenimi i acideve karboksilike alifatike.
Acidet karboksilike alifatike halogjenohen në pozicionin α me klor ose brom në prani të sasive katalitike fosfori i kuq ose halidet e fosforit (Reagimi Gell-Volhard-Zelinsky ). Për shembull, kur acidi heksanoik brominohet në prani të fosforit të kuq ose klorurit të fosforit (III), acidi 2-bromoheksanoik formohet me rendiment të lartë, për shembull:
Nuk është vetë acidi karboksilik që i nënshtrohet brominimit, por kloruri acid i formuar prej tij në vend. Kloruri i acidit ka veti më të forta të acidit CH sesa acidi karboksilik dhe më lehtë formon formën e enolit.
Enoli (I) shton bromin për të formuar një derivat halogjen (II), i cili më tej abstraksionon një halogjen hidrogjeni dhe kthehet në një halogjen acidi të zëvendësuar me α-halogjen (III). Në fazën e fundit, halidi i acidit karboksilik i pazëvendësuar rigjenerohet.

Acidet e tjera heterofunksionale sintetizohen nga acidet α-halo-zëvendësuese që rezultojnë duke përdorur reaksionet e zëvendësimit nukleofilik.
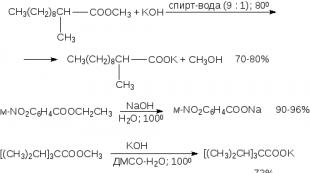
Hidroliza e estereve katalizohet si nga acidet ashtu edhe nga bazat. Hidroliza acide e estereve zakonisht kryhet duke ngrohur me acid klorhidrik ose sulfurik në një mjedis ujor ose ujor-alkoolik. Në sintezën organike, hidroliza acide e estereve përdoret më shpesh për esteret malonikë të zëvendësuar me mono- dhe dialkil (Kapitulli 17). Derivatet mono- dhe të dyzëvendësuar të esterit malonik, kur zihen me acid klorhidrik të përqendruar, i nënshtrohen hidrolizës së ndjekur nga dekarboksilimi.
Për hidrolizën e katalizuar me bazë, zakonisht përdoret një zgjidhje ujore ose ujore-alkoolike e NaOH ose KOH. Rezultatet më të mira merren duke përdorur një suspension të hollë të hidroksidit të kaliumit në DMSO që përmban një sasi të vogël uji.

Metoda e fundit preferohet për saponifikimin e estereve të acideve të penguara, një modifikim tjetër i kësaj metode është hidroliza alkaline e estereve të penguar në prani të poliesterit 18-kurorë-6:

Për qëllime përgatitore, hidroliza e katalizuar me bazë ka një numër avantazhesh të qarta mbi hidrolizën acidike. Shpejtësia e hidrolizës bazë të estereve është zakonisht një mijë herë më e shpejtë se ajo e katalizimit acid. Hidroliza në një mjedis acid është një proces i kthyeshëm, në ndryshim nga hidroliza në prani të një baze, e cila është e pakthyeshme.
18.8.2.A. Mekanizmat e hidrolizës së esterit
Hidroliza e estereve me ujë të pastër është në shumicën e rasteve një reaksion i kthyeshëm, duke çuar në një përzierje ekuilibri të acidit karboksilik dhe esterit fillestar:

Ky reaksion në mjediset acidike dhe alkaline përshpejtohet shumë, gjë që shoqërohet me katalizën acido-bazike (Kapitulli 3).
Sipas K. Ingold, mekanizmat e hidrolizës së esterit klasifikohen sipas kritereve të mëposhtme:
(1) Lloji i katalizës: acid (simboli A) ose bazik (simboli B);
(2) Lloji i ndarjes, që tregon se cila nga dy lidhjet -C-O në ester është shkëputur si rezultat i reaksionit: oksigjen acil (indeksi AC) ose oksigjen alkil (indeksi AL):

(3) Molekulariteti i reaksionit (1 ose 2).
Nga këto tre kritere mund të bëhen tetë kombinime të ndryshme, të cilat janë paraqitur në figurën 18.1.

Këto janë mekanizmat më të zakonshëm. Saponifikimi alkalik është pothuajse gjithmonë i tipit B AC 2. Hidroliza e acidit (si dhe esterifikimi) në shumicën e rasteve ka një mekanizëm A AC 2.

Mekanizmi AAC 1 zakonisht vërehet vetëm në solucione me aciditet të fortë (për shembull, në konk. H2SO 4), dhe është veçanërisht i zakonshëm për esteret e acideve aromatike të penguara sterikisht.
Mekanizmi i BAC 1 është ende i panjohur.

Mekanizmi BAL 2 u gjet vetëm në rastin e grupeve acil jashtëzakonisht të forta hapësinore dhe hidrolizës neutrale të -laktoneve. Mekanizmi i A AL 2 është ende i panjohur.

Sipas mekanizmit Dhe AL 1 zakonisht reagojnë esteret terciar-alkil në një mjedis neutral ose acid. Të njëjtat nënshtresa në kushte të ngjashme mund të reagojnë sipas mekanizmit B AL 1, megjithatë, me kalimin në një mjedis pak më alkalik, mekanizmi BAL 1 zëvendësohet menjëherë nga mekanizmi B AC 2.
Siç mund të shihet nga Skema 18.1, reaksionet e katalizuara nga acidet janë të kthyeshme dhe nga parimi i kthyeshmërisë mikroskopike (Kapitulli 2) rezulton se esterifikimi i katalizuar nga acidi gjithashtu zhvillohet me mekanizma të ngjashëm. Megjithatë, me katalizën bazë, ekuilibri zhvendoset drejt hidrolizës (saponifikimi), pasi ekuilibri zhvendoset për shkak të jonizimit të acidit karboksilik. Sipas skemës së mësipërme, në rastin e mekanizmit A AC 1, grupet COOR dhe COOH protonohen në atomin e oksigjenit alkoksi ose hidroksil. Në përgjithësi, nga pikëpamja e termodinamikës, protonizimi i oksigjenit karbonil, grupi C=O, është më i favorshëm, sepse në këtë rast, ngarkesa pozitive mund të delokalizohet midis të dy atomeve të oksigjenit:

Megjithatë, tretësira përmban edhe një kation tautomerik, një ndërmjetës i domosdoshëm në mekanizmin A AC 1, në sasi të vogla. Të dy mekanizmat B1 (prej të cilëve B AC 1 është i panjohur) në fakt nuk janë fare katalitik, sepse në fillim shpërbërja i eterit neutral ndodh.
Nga tetë mekanizmat Ingold, vetëm gjashtë janë provuar eksperimentalisht.
Hidroliza e estereve katalizohet si nga acidet ashtu edhe nga bazat. Hidroliza acide e estereve zakonisht kryhet duke ngrohur me acid klorhidrik ose sulfurik në një mjedis ujor ose ujor-alkoolik. Në sintezën organike, hidroliza acide e estereve përdoret më shpesh për esteret malonikë të zëvendësuar me mono- dhe dialkil (Kapitulli 17). Derivatet mono- dhe të dyzëvendësuar të esterit malonik, kur zihen me acid klorhidrik të përqendruar, i nënshtrohen hidrolizës së ndjekur nga dekarboksilimi.
Për hidrolizën e katalizuar me bazë, zakonisht përdoret një zgjidhje ujore ose ujore-alkoolike e NaOH ose KOH. Rezultatet më të mira merren duke përdorur një suspension të hollë të hidroksidit të kaliumit në DMSO që përmban një sasi të vogël uji.

Metoda e fundit preferohet për saponifikimin e estereve të acideve të penguara, një modifikim tjetër i kësaj metode është hidroliza alkaline e estereve të penguar në prani të poliesterit 18-kurorë-6:

Për qëllime përgatitore, hidroliza e katalizuar me bazë ka një numër avantazhesh të qarta mbi hidrolizën acidike. Shpejtësia e hidrolizës bazë të estereve është zakonisht një mijë herë më e shpejtë se ajo e katalizimit acid. Hidroliza në një mjedis acid është një proces i kthyeshëm, në ndryshim nga hidroliza në prani të një baze, e cila është e pakthyeshme.
18.8.2.A. Mekanizmat e hidrolizës së esterit
Hidroliza e estereve me ujë të pastër është në shumicën e rasteve një reaksion i kthyeshëm, duke çuar në një përzierje ekuilibri të acidit karboksilik dhe esterit fillestar:

Ky reaksion në mjediset acidike dhe alkaline përshpejtohet shumë, gjë që shoqërohet me katalizën acido-bazike (Kapitulli 3).
Sipas K. Ingold, mekanizmat e hidrolizës së esterit klasifikohen sipas kritereve të mëposhtme:
(1) Lloji i katalizës: acid (simboli A) ose bazik (simboli B);
(2) Lloji i ndarjes, që tregon se cila nga dy lidhjet -C-O në ester është shkëputur si rezultat i reaksionit: oksigjen acil (indeksi AC) ose oksigjen alkil (indeksi AL):

(3) Molekulariteti i reaksionit (1 ose 2).
Nga këto tre kritere mund të bëhen tetë kombinime të ndryshme, të cilat janë paraqitur në figurën 18.1.

Këto janë mekanizmat më të zakonshëm. Saponifikimi alkalik është pothuajse gjithmonë i tipit B AC 2. Hidroliza e acidit (si dhe esterifikimi) në shumicën e rasteve ka një mekanizëm A AC 2.

Mekanizmi AAC 1 zakonisht vërehet vetëm në solucione me aciditet të fortë (për shembull, në konk. H2SO 4), dhe është veçanërisht i zakonshëm për esteret e acideve aromatike të penguara sterikisht.
Mekanizmi i BAC 1 është ende i panjohur.

Mekanizmi BAL 2 u gjet vetëm në rastin e grupeve acil jashtëzakonisht të forta hapësinore dhe hidrolizës neutrale të -laktoneve. Mekanizmi i A AL 2 është ende i panjohur.

Sipas mekanizmit Dhe AL 1 zakonisht reagojnë esteret terciar-alkil në një mjedis neutral ose acid. Të njëjtat nënshtresa në kushte të ngjashme mund të reagojnë sipas mekanizmit B AL 1, megjithatë, me kalimin në një mjedis pak më alkalik, mekanizmi BAL 1 zëvendësohet menjëherë nga mekanizmi B AC 2.
Siç mund të shihet nga Skema 18.1, reaksionet e katalizuara nga acidet janë të kthyeshme dhe nga parimi i kthyeshmërisë mikroskopike (Kapitulli 2) rezulton se esterifikimi i katalizuar nga acidi gjithashtu zhvillohet me mekanizma të ngjashëm. Megjithatë, me katalizën bazë, ekuilibri zhvendoset drejt hidrolizës (saponifikimi), pasi ekuilibri zhvendoset për shkak të jonizimit të acidit karboksilik. Sipas skemës së mësipërme, në rastin e mekanizmit A AC 1, grupet COOR dhe COOH protonohen në atomin e oksigjenit alkoksi ose hidroksil. Në përgjithësi, nga pikëpamja e termodinamikës, protonizimi i oksigjenit karbonil, grupi C=O, është më i favorshëm, sepse në këtë rast, ngarkesa pozitive mund të delokalizohet midis të dy atomeve të oksigjenit:

Megjithatë, tretësira përmban edhe një kation tautomerik, një ndërmjetës i domosdoshëm në mekanizmin A AC 1, në sasi të vogla. Të dy mekanizmat B1 (prej të cilëve B AC 1 është i panjohur) në fakt nuk janë fare katalitik, sepse në fillim shpërbërja i eterit neutral ndodh.
Nga tetë mekanizmat Ingold, vetëm gjashtë janë provuar eksperimentalisht.