Marrja e hekurit dhe sinteza e hemoglobinës. Anemitë e shkaktuara nga çrregullimet e sintezës së hemoglobinës dhe metabolizmit të hekurit Lipoliza dhe lipogjeneza. Kuptimi. Varësia e lipogjenezës nga ritmi ushqyes dhe përbërja e ushqimit. Rregullimi i lipolizës dhe lipogjenezës. Transporti dhe përdorimi
Sinteza e kompleksit të pirrolit në trup rrjedh nga pararendësit me peshë të ulët molekulare de novo. Burimet e hekurit janë ushqimet dhe hekuri, i cili çlirohet gjatë prishjes së rruazave të kuqe të gjakut.
Sinteza e hemit.
Unëfazë... Glicina dhe succinyl-CoA janë të përfshira. 5-aminolevulinate sintaza- një enzimë kyçe alosterike për sintezën e tetrapirrolit. Koenzima - piridoksal fosfat. I nxitur nga steroidet dhe i frenuar nga reagimet nga produkti përfundimtar, hemi. Formuar Acidi 5-aminolevulinik(-ALK).
IIfazë... Arsimi porfobilinogjen PBG. Enzimë porfobilinogjen sintaza frenohet nga produktet përfundimtare të sintezës.
IIIfazë... Shumëfazësh. Kompleksi tetrapirroli sintetizohet nga 4 molekula porfobilinogjene protoporfirinaIX.
IVfazë... Protoporfirina IX lidh hekurin me pjesëmarrjen ferrokelataza (hemsintaza), dhe u formua heme... Burimi i hekurit është ferritina. Vitamina B 12 dhe jonet e bakrit janë të përfshirë në sintezën e hemit.
Pjesa e proteinave molekula e hemoglobinës sintetizohet në të njëjtën mënyrë si të gjitha proteinat e tjera. Sinteza e zinxhirëve polipeptidikë të hemoglobinës ndodh vetëm në prani të hemit.
2.7. Shkëmbimi i nukleoproteinave
Rënia e NK. Nën ndikimin e enzimave të stomakut, pjesërisht të acidit klorhidrik, nukleoproteinat e ushqimit shpërbëhen në polipeptide dhe NK. Zbërthimi i NK ndodh në zorrën e hollë në mënyrë hidrolitike nën veprimin e nukleaza lëng pankreatik. Ato i përkasin fosfodiesterazave. ekziston endonukleazat dhe ekzonukleazë, ribonukleazë dhe deoksiribonukleaza. Produktet e hidrolizës janë mononukleotide dhe oligonukleotide. Nukleazat çajnë molekulat e NK në inde.
Zbërthimi i fosfateve nukleozide. Faza e parë është eliminimi i mbetjeve të acidit fosforik. Në fazën e dytë, kariboza e mbetur transferohet nga nukleozidi në acidin fosforik. Ky reagim po përshpejtohet riboziltransferazat.
F-U-A F + U-A; U-A + F U-F + A
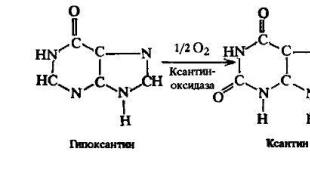
Zbërthimi i bazave purine fillon me deaminimin e atyre që kanë amino grupe. Përfshihen aminohidrolaza specifike.
Adenina hipoksantina; guaninë ksantinë
Hipoksantina dhe ksantina oksidohet në acid urik, enzima - ksantine oksidaza.

Acidi urik prodhohet kryesisht në mëlçi. Është produkti kryesor i katabolizmit të nukleotideve të purinës tek njerëzit. Në trup, formon 0,5-1 g në ditë, ekskretohet përmes veshkave. Rritja kronike e përqendrimit të acidit urik ( hiperuricemia) shpesh çon në zhvillim përdhes... Kriza e përdhes shoqërohet me depozitimin e kristaleve të uratit të natriumit në kyç. Hiperuricemia është zakonisht e trashëgueshme.
Zbërthimi i pirimidinës baza gjithashtu fillon me deaminimin. Baza pirimidine të deaminuara janë duke u restauruar. Acidi karbamik dhe-alanina janë produktet përfundimtare të zbërthimit të U dhe C. Nga T në vend të alaninës, formohet acidi -aminoizobutirik.
 …
…
Sinteza e nukleotideve të pirimidinës y, c, t
Nga CO 2 , gln, asp të sintetizuara acid monofosforik uridin... Ai shërben si një pararendës i nukleotideve citidil dhe timidil pirimidine.
Reagimi i parë është formimi i karbamoil fosfatit nën veprimin e karbamoil fosfat sintetazës II (e varur nga glutamati, që gjendet në citosol).
CO 2 + Glutaminë + 2 ATP + H 2 O N 2 N CO ORO 3 N 2 + 2 ADP + H 3 RO 4 .
Karbamoil fosfati më pas reagon me aspartat. Si rezultat i një sërë reaksionesh, formohet acidi uridil.
Orotaciduria- ekskretimi i sasive të mëdha të acidit orotik në urinë. Orotaciduria e njohur trashëgimore, në të cilën lirohet deri në 1.5 g acid orotik në ditë, 1000 herë më shumë se normalja. Sëmundja shoqërohet me mungesë të një enzime që katalizon formimin dhe dekarboksilimin e acidit orotidilik. Orotaciduria e trashëguar çon në zhvillimin e një vonese të mprehtë të pakthyeshme në zhvillimin mendor dhe fizik; zakonisht pacientët vdesin në vitet e para të jetës. Acidi orotik nuk është toksik; Çrregullimet e zhvillimit janë rezultat i “urisë pirimidinike”. Prandaj, uridina përdoret për të trajtuar këtë sëmundje.
Reduktimi - dhuruesi i hidrogjenit - proteina tioredoksina që përmban grupe SH;
Aminimi - burimi i grupit amino është gln;
- Burimi i metilimit të grupit metil - metilen H 4 -folat.
Kur ndërveprojnë me ATP, sintetizohen trifosfatet e lira pirimidine nukleozide.

Sinteza e eritrociteve- një nga proceset më të fuqishme të formimit të qelizave në trup. Normalisht, rreth 2 milionë qeliza të kuqe të gjakut formohen çdo sekondë, 173 miliardë në ditë dhe 63 trilionë në vit. Nëse këto vlera i përkthejmë në masë, atëherë çdo ditë formohen rreth 140 g eritrocite, çdo vit - 51 kg, dhe masa e eritrociteve të formuara në trup mbi 70 vjet është rreth 3.5 ton.
Në një të rritur eritropoeza paraqitet në palcën e eshtrave të kockave të sheshta, ndërsa tek fetusi gjenden ishujt e hematopoiezës në mëlçi dhe shpretkë (hematopoieza ekstramedulare). Në disa gjendje patologjike (talasemia, leucemia, etj.), tek një i rritur mund të gjenden vatra të hematopoiezës ekstramedulare.
Një nga elementët e rëndësishëm të ndarjes qelizore është vitaminë B e nevojshme për sintezën e ADN-së, duke qenë, në fakt, një katalizator për këtë reagim. Në procesin e sintezës së ADN-së, vitamina B12 nuk konsumohet, por në mënyrë ciklike hyn në reaksione si një substancë aktive; si rezultat i këtij cikli, nga monofosfati i uridinës formohet monofosfati i timidinës. Me një ulje të nivelit të vitaminës B12, uridina inkorporohet dobët në molekulën e ADN-së, gjë që çon në çrregullime të shumta, në veçanti, maturim të dëmtuar të qelizave të gjakut.
Një faktor tjetër që ndikon në ndarjen e qelizave është acid folik... Ajo, si një koenzimë, në veçanti, është e përfshirë në sintezën e nukleotideve purine dhe pirimidine.
Skema e përgjithshme e hematopoiezës postembrionale
Hematopoeza(hematopoiesis) është një sistem shumë dinamik, i ekuilibruar mirë, që rinovohet vazhdimisht. Paraardhësi i vetëm i hematopoiezës është qeliza staminale. Sipas koncepteve moderne, kjo është një klasë e tërë qelizash të vendosura në ontogjenezë, vetia kryesore e së cilës është aftësia për të dhënë të gjitha mikrobet hematopoietike - eritrocitike, megakariocitare, granulocitare (eozinofile, bazofile, neutrofile), monocito-makrofagë. , T-limfocitare, B-limfocitare.
Si rezultat i disa ndarjeve, qelizat humbasin aftësinë e tyre për të qenë paraardhës universal dhe kthehen në qeliza pluripotente. E tillë, për shembull, është qeliza pararendëse e mielopezës (eritrocitet, megakariocitet, granulocitet). Pas disa ndarjeve të tjera, pas universalitetit, zhduket edhe pluripotenca, qelizat bëhen unipotente (uniˮ është e vetmja), domethënë janë të afta të diferencohen vetëm në një drejtim.

Qelizat më të ndara në palcën e eshtrave janë qelizat pararendëse të mielopezës (shih figurën ⭡); ndërsa diferencimi vazhdon, numri i ndarjeve të mbetura zvogëlohet dhe qelizat e kuqe të gjakut të dallueshme morfologjikisht ndalojnë gradualisht ndarjen.
Diferencimi i qelizave eritroide
Vetë linja qelizore eritroide (erythron) fillon me qeliza unipotente që formojnë shpërthime, të cilat janë pasardhës të qelizave paraardhëse të mielopezës. Qelizat që formojnë shpërthim në kulturën e indeve rriten në koloni të vogla që i ngjajnë një shpërthimi (shpërthimi). Për maturimin e tyre, kërkohet një ndërmjetës i veçantë - aktiviteti nxitës i shpërthimit. Ky është një faktor i ndikimit të mikromjedisit në qelizat e pjekura, një faktor i ndërveprimit ndërqelizor.
Ekzistojnë dy popullata të qelizave që formojnë shpërthime: e para rregullohet ekskluzivisht nga aktiviteti nxitës i shpërthimit, e dyta bëhet e ndjeshme ndaj efekteve të eritropoietinës. Në popullsinë e dytë fillon sinteza e hemoglobinës duke vazhduar në qelizat e ndjeshme ndaj eritropoietinës dhe në qelizat pasuese të maturimit.
Në fazën e qelizave që formojnë shpërthim, ndodh një ndryshim thelbësor në aktivitetin qelizor - nga ndarja në sintezën e hemoglobinës. Në qelizat e mëvonshme, ndarja pezullohet (qeliza e fundit në këtë rresht e aftë për ndarje është eritroblasti polikromatofil), bërthama zvogëlohet në madhësi absolute dhe në raport me vëllimin e citoplazmës, në të cilën bëhet sinteza e substancave. Në fazën e fundit, bërthama hiqet nga qeliza, pastaj mbetjet e ARN zhduken; ende mund të gjenden me një njollë të veçantë në eritrocitet e reja - retikulocitet, por nuk mund të gjenden në eritrocitet e pjekura.
Skema e fazave kryesore të diferencimit të qelizave eritroide është si më poshtë:
qeliza staminale pluripotente ⭢ njësi shpërthyese e serisë eritroide (BFU-E) ⭢ njësi koloni-formuese e serisë eritroide (CFU-E) ⭢ eritroblast ⭢ pronormocit ⭢ normocit bazofilik ⭢ normocit bazofilik.
Rregullimi i eritropoezës
Proceset e rregullimit të hematopoiezës ende nuk janë studiuar mjaftueshëm. Nevoja për të ruajtur vazhdimisht hematopoiezën, për të kënaqur në mënyrë adekuate nevojat e trupit në qeliza të ndryshme të specializuara, për të siguruar qëndrueshmërinë dhe ekuilibrin e mjedisit të brendshëm (homeostaza) - e gjithë kjo presupozon ekzistencën e mekanizmave rregullues kompleksë që veprojnë në parimin e reagimit.
Faktori humoral më i famshëm në rregullimin e eritropoezës është hormoni eritropoietin... Është një faktor stresi që sintetizohet në qeliza të ndryshme dhe në organe të ndryshme. Pjesa më e madhe e saj formohet në veshka, megjithatë, edhe në mungesë të tyre, eritropoetina prodhohet nga endoteli vaskular dhe mëlçia. Niveli i eritropoetinës është i qëndrueshëm dhe ndryshon në rritje me humbje të mprehtë dhe të bollshme gjaku, hemolizë akute, kur ngjiteni në male, me ishemi akute renale. Në mënyrë paradoksale, në aneminë kronike, nivelet e eritropoietinës janë zakonisht normale, me përjashtim të anemisë aplastike, ku niveli i saj është vazhdimisht jashtëzakonisht i lartë.
Së bashku me eritropoietinën, në gjak janë të pranishëm edhe frenuesit e eritropoezës. Ky është një numër i madh substancash të ndryshme, disa prej të cilave mund t'i atribuohen toksinave molekulare mesatare që grumbullohen si rezultat i proceseve patologjike që lidhen me formimin e tyre të shtuar ose dëmtimin e sekretimit të tyre.
Në fazat e hershme të diferencimit, rregullimi i eritronit kryhet kryesisht për shkak të faktorëve të mikromjedisit qelizor, dhe më vonë - me një ekuilibër të aktivitetit të eritropoetinës dhe frenuesve të eritropoezës. Në situata akute, kur është e nevojshme të krijohet shpejt një numër i madh eritrocitesh të reja, aktivizohet mekanizmi i eritropoietinës së stresit - një mbizotërim i mprehtë i aktivitetit të eritropoetinës mbi aktivitetin e frenuesve eritropoetikë. Në situata patologjike, përkundrazi, aktiviteti frenues mund të mbizotërojë mbi eritropoietinën, gjë që çon në frenimin e eritropoezës.
Sinteza e hemoglobinës
Hemoglobina përmban hekur. Një sasi e pamjaftueshme e këtij elementi në trup mund të çojë në zhvillimin e anemisë (shih. Anemi nga mungesa e hekurit). Ekziston një lidhje midis aftësisë për të sintetizuar një sasi të caktuar të hemoglobinës (që është për shkak të rezervave të hekurit) dhe eritropoezës - sipas të gjitha gjasave, ekziston një vlerë pragu e përqendrimit të hemoglobinës, pa të cilën eritropoeza ndalon.
Sinteza e hemoglobinës fillon në prekursorët eritroide në fazën e formimit të një qelize të ndjeshme ndaj eritropoietinës. Në fetus, dhe më pas në periudhën e hershme pas lindjes, fëmija zhvillon hemoglobinën F, dhe më pas, kryesisht hemoglobinën A. Kur eritropoeza është e stresuar (hemolizë, gjakderdhje), një sasi e caktuar e hemoglobinës F mund të shfaqet në gjakun e një të rrituri. .
Hemoglobina përbëhet nga dy variante të zinxhirëve të globinës, a dhe p, që rrethojnë hemin që përmban hekur. Në varësi të ndryshimeve në sekuencat e mbetjeve të aminoacideve në zinxhirët e globinës, vetitë kimike dhe fizike të hemoglobinës ndryshojnë, në kushte të caktuara ajo mund të kristalizohet dhe të bëhet e pazgjidhshme (për shembull, hemoglobina S në aneminë drapërocitare).
Vetitë e rruazave të kuqe të gjakut
Qelizat e kuqe të gjakut kanë disa veti. Më i njohuri është transporti i oksigjenit (O2) dhe dioksidit të karbonit (CO2). Ajo kryhet nga hemoglobina, e cila lidhet në mënyrë alternative me njërin dhe tjetrin gaz, në varësi të tensionit të gazit përkatës në mjedis: në mushkëri - oksigjen, në inde - dioksid karboni. Kimia e reaksionit konsiston në zhvendosjen dhe zëvendësimin e një gazi me një tjetër nga lidhja me hemoglobinën. Përveç kësaj, eritrocitet janë bartës të oksidit nitrik (NO), i cili është përgjegjës për tonin e enëve të gjakut dhe gjithashtu është i përfshirë në sinjalizimin e qelizave dhe shumë procese të tjera fiziologjike.
Eritrocitet kanë aftësinë të ndryshojnë formën e tyre kur kalojnë nëpër kapilarët me diametër të vogël. Qelizat përhapen, rrotullohen në një spirale. Plasticiteti i eritrociteve varet nga faktorë të ndryshëm, duke përfshirë strukturën e membranës së eritrociteve, llojin e hemoglobinës që përmban dhe citoskeletin. Përveç kësaj, membrana e eritrociteve është e rrethuar nga një lloj "re" e proteinave të ndryshme që mund të ndryshojnë deformueshmërinë. Këto përfshijnë komplekset imune, fibrinogjen. Këto substanca ndryshojnë ngarkesën e membranës së eritrociteve, lidhen me receptorët dhe përshpejtojnë sedimentimin e eritrociteve në një kapilar qelqi.
Në rastin e formimit të trombit, eritrocitet janë qendrat e formimit të fijeve të fibrinës, kjo jo vetëm që mund të ndryshojë deformueshmërinë, të shkaktojë grumbullimin e tyre, ngjitjen në kolonat e monedhave, por edhe të copëtojë qelizat e kuqe të gjakut në fragmente, të heqë copa membranash prej tyre.
Reaksioni i sedimentimit të eritrociteve (ESR) pasqyron praninë në sipërfaqen e tyre të një ngarkese që largon eritrocitet nga njëri-tjetri. Shfaqja gjatë reaksioneve inflamatore, me aktivizim të koagulimit etj. një re dielektrike rreth eritrocitit çon në një ulje të forcave repulsive, si rezultat i së cilës eritrocitet fillojnë të vendosen më shpejt në një kapilar të vendosur vertikalisht. Nëse kapilari është i anuar 45 °, atëherë forcat refuzuese veprojnë vetëm gjatë kalimit të eritrociteve nëpër lumenin e kapilarit. Kur qelizat arrijnë në mur, ato rrokullisen poshtë tij pa hasur në rezistencë. Si rezultat, në një kapilar të anuar, shkalla e sedimentimit të eritrociteve rritet dhjetëfish.
Burimet:
1. Sindroma anemike në praktikën klinike / P.А. Vorobiev, - M., 2001;
2. Hematologjia: Libri më i fundit referues / Ed. K.M. Abdulkadyrov. - M., 2004.
1. Funksionet e qelizave të gjakut. Funksionet e eritrociteve. Vetitë e eritrociteve. Cikli Embden-Meyerhof. Struktura e eritrociteve.
2. Hemoglobina. Llojet (llojet) e hemoglobinës. Sinteza e hemoglobinës. Funksioni i hemoglobinës. Struktura e hemoglobinës.
3. Plakja e eritrociteve. Shkatërrimi i qelizave të kuqe të gjakut. Jetëgjatësia e një eritrociti. Ekinociti. Ekinocitet.
4. Hekuri. Hekuri është normal. Roli i joneve të hekurit në eritropoezë. Transferrinë. Nevoja e trupit për hekur. Mungesa e hekurit. OZHSS.
5. Eritropoeza. Ishujt eritroblastike. Anemia. Eritrocitoza.
6. Rregullimi i eritropoezës. Eritropoietina. Hormonet seksuale dhe eritropoeza.
7. Leukocitet. Leukocitoza. Leukopenia. Granulocitet. Formula e leukociteve.
8. Funksionet e granulociteve neutrofile (leukociteve). Mbrojtjet. Katelicidinat. Proteinat e fazës akute. Faktorët kemotaktikë.
9. Efekti baktericid i neutrofileve. Granulopoeza. Granulopoeza neutrofile. Granulocitoza. Neutropenia.
10. Funksionet e bazofileve. Funksionet e granulociteve bazofile. Sasi normale. Histamine. Heparina.
Hemoglobina. Llojet (llojet) e hemoglobinës. Sinteza e hemoglobinës. Funksioni i hemoglobinës. Struktura e hemoglobinës.
Hemoglobinaështë një hemoproteinë, me peshë molekulare rreth 60 mijë, e cila e njollos eritrocitin me ngjyrë të kuqe pas lidhjes së molekulës O2 me jonin e hekurit (Fe ++). Kanë burra 1 litër gjak përmban 157 (140-175) g hemoglobina, në femrat- 138 (123-153) g. Molekula e hemoglobinës përbëhet nga katër nënnjësi heme të lidhura me pjesën proteinike të molekulës - globina e formuar nga vargje polipeptide.
Sinteza e hemit vazhdon në mitokondritë e eritroblasteve. Sinteza e zinxhirit të globinës kryhet në poliribozome dhe kontrollohet nga gjenet e kromozomeve të 11-të dhe të 16-të. Skema e sintezës së hemoglobinës tek njerëzit është paraqitur në Fig. 7.2.
Hemoglobina që përmban dy zinxhirë a dhe dy B quhet tipi A (nga i rritur - i rritur). 1 g hemoglobinë të tipit A lidh 1,34 ml O2. Në tre muajt e parë të jetës së një fetusi njerëzor, gjaku përmban hemoglobina embrionale të tipit Gower I (4 zinxhirë epsilon) dhe Gower II (zinxhirët 2a dhe 25). Më pas u formua hemoglobina F(nga fetus - fruta). Globina e saj përfaqësohet nga dy zinxhirë a dhe dy B. Hemoglobina F ka afinitet 20-30% më të lartë për O2 sesa hemoglobina A, e cila kontribuon në një furnizim më të mirë të fetusit me oksigjen. Në lindjen e një fëmije paraqitet deri në 50-80% e hemoglobinës hemoglobina F dhe 15-40% - lloji A, dhe deri në 3 vjet niveli hemoglobina F zvogëlohet në 2%.
Komponimi i hemoglobinës me një molekulë 02 quhet oksihemoglobinë. Afiniteti i hemoglobinës ndaj oksigjenit dhe shkëputja e oksihemoglobinës (shkëputja e molekulave të oksigjenit nga oksihemoglobina) varet nga tensioni i oksigjenit (PO2), dioksidi i karbonit (PCO2) në gjak, pH e gjakut, temperatura e gjakut dhe përqendrimi i 2,3-DPG në eritrocite. . Pra, afiniteti rritet me një rritje të PO2 ose një ulje të PCO2 në gjak, një shkelje e formimit të 2,3-DPG në eritrocite. Përkundrazi, një rritje në përqendrimin e 2,3-DPG, një rënie në PO2 në gjak, një zhvendosje e pH në anën acidike, një rritje e PCO2 dhe temperaturës së gjakut - zvogëlon afinitetin e hemoglobinës për oksigjenin, duke lehtësuar kështu lëshimi në inde. 2,3-DPG lidhet me zinxhirët β hemoglobina, duke lehtësuar shkëputjen e 02 nga molekula e hemoglobinës.
Një rritje e përqendrimit të 2,3-DPG vërehet te njerëzit e trajnuar për punë fizike afatgjatë, të përshtatur për një qëndrim të gjatë në male. Oksihemoglobina që hoqi dorë nga oksigjeni quhet oksigjen i reduktuar, ose deoksihemoglobina... Në një gjendje pushimi fiziologjik tek njerëzit, hemoglobina në gjakun arterial është 97% e ngopur me oksigjen, në gjakun venoz - me 70%. Sa më i theksuar të jetë konsumi i oksigjenit nga indet, aq më i ulët është ngopja me oksigjen në gjakun venoz. Për shembull, me punë intensive fizike, konsumi i oksigjenit nga indet e muskujve rritet disa dhjetëra herë dhe ngopja e oksigjenit e gjakut venoz që rrjedh nga muskujt zvogëlohet në 15%. Përmbajtja e hemoglobinës në një eritrocit të vetëmështë 27,5-33,2 pikogramë. Një rënie në këtë vlerë tregon hipokromik(d.m.th., ulur), rrit - o hiperkromik(dmth rritur) përmbajtjen e hemoglobinës në eritrocite. Ky tregues ka vlerë diagnostikuese. Për shembull, hiperkromi eritrocitare karakteristikë e anemisë së mungesës B | 2, hipokromi- për aneminë e mungesës së hekurit.
Porfiritë - një grup heterogjen sëmundjesh të shkaktuara nga sinteza e dëmtuar e hemit për shkak të mungesës së një ose më shumë enzimave.
Klasifikimi i porfirisë
Nuk ka një klasifikim uniform të porfirisë. Porfiritë ndahen për arsye:
E trashëgueshme... Ato lindin me një defekt në gjenin e enzimës së përfshirë në sintezën e hemit;
I fituar... Ato lindin me efektin frenues të përbërjeve toksike (heksoklorobenzen, kripërat e metaleve të rënda - plumbi) në enzimat e sintezës së hemit.
Në varësi të lokalizimit të preferuar të mungesës së enzimës (në mëlçi ose në eritrocite), porfirina ndahet në:
hepatike- lloji më i zakonshëm i porfirinës është porfiria akute intermitente (AKI), porfiria e vonshme e lëkurës, koproporfiria trashëgimore, porfiria mozaike;
eritropoetike- porfiria eritropoetike kongjenitale (sëmundja e Gunther-it), protoporfiria eritropoetike.
Në varësi të pamjes klinike, porfiritë ndahen në:
kronike.
Pasojat negative të porfirisë shoqërohen me mungesë të hemit dhe akumulimin në inde dhe gjak të produkteve të ndërmjetme të sintezës së hemit - porfirinogjeneve dhe produkteve të tyre të oksidimit. Në porfiritë eritropoetike, porfirinat grumbullohen në normoblaste dhe eritrocite, në porfiritë hepatike - në hepatocitet.
Për çdo lloj porfirie, ekziston një nivel i caktuar i defektit enzimatik, si rezultat, produktet e sintetizuara mbi këtë nivel grumbullohen. Këto ushqime janë shënuesit kryesorë diagnostikues të sëmundjes.
Porfirinogjenët janë helmues; në format e rënda të porfirisë, ato shkaktojnë çrregullime neuropsikiatrike, mosfunksionim të RES dhe dëmtime të lëkurës.
Çrregullimet neuropsikiatrike në porfiritë shoqërohen me faktin se aminolevulinati dhe porfirinogjenët janë neurotoksina.
Në lëkurë, në diell, porfirinogjenët konvertohen lehtësisht në porfirina. Oksigjeni, kur ndërvepron me porfirinat, kalon në një gjendje të vetme. Oksigjeni i vetëm stimulon peroksidimin e lipideve të membranave qelizore dhe shkatërrimin e qelizave; prandaj, porfiritë shpesh shoqërohen me fotosensibilizimin dhe ulçerimin e zonave të hapura të lëkurës.
Porfirinogjenët nuk janë as me ngjyrë dhe as fluoreshente, ndërsa porfirinat shfaqin fluoreshencë intensive të kuqe nën dritën ultravjollcë. Porfirinat e tepërta, të cilat ekskretohen në urinë, i japin një ngjyrë të errët ("porfirina" në greqisht do të thotë vjollcë).
Ndonjëherë, me forma të lehta të porfirisë trashëgimore, sëmundja mund të jetë asimptomatike, por marrja e barnave që nxisin sintezën e aminolevulinat sintazës mund ta përkeqësojë sëmundjen. Në disa raste, simptomat e sëmundjes nuk shfaqen deri në pubertet, kur një rritje në formimin e β-steroideve shkakton sintezën e aminolevulinat sintazës. Porfiritë vërehen edhe në rast të helmimit me kripëra plumbi, pasi plumbi frenon aminolevulinate dehidratazën dhe ferrokelatazën. Disa herbicide dhe insekticide të halogjenizuara janë nxitës të sintezës së aminolevulinat sintazës, kështu që gëlltitja e tyre shoqërohet me simptoma të porfirisë.
Llojet e porfirive
Porfiria akute intermitente (AKI) - shkak - një defekt në gjenin që kodon PBG - deaminaza. Trashëgohet në mënyrë autosomale dominante. Ekziston një grumbullim i prekursorëve të hershëm të sintezës së hemit: 5-ALA (5-ALA) dhe porfobilinogjen (PBG).
PBG pa ngjyrë në dritë shndërrohet në porfibilinë dhe porfirinë, ato e bëjnë urinën të errët. ALA ka një efekt neurotoksik, duke çuar në paralizë të dobët të gjymtyrëve dhe parezë të muskujve të frymëmarrjes. Kjo e fundit shkakton insuficiencë respiratore akute. Sëmundja manifestohet në moshën e mesme, provokohet nga marrja e analgjezikëve, barnave sulfa, pasi ato rrisin sintezën e ALA - sintazës.
Simptomat klinike janë dhimbje akute abdominale, të vjella, kapsllëk, çrregullime kardiovaskulare, çrregullime neuropsikiatrike. Nuk ka mbindjeshmëri ndaj dritës, pasi çrregullimi metabolik ndodh në fazën që i paraprin formimit të uroporfirinogjenit.
Për trajtim, përdoret ilaçi normosang - heme arginate. Veprimi bazohet në faktin se hemi, nëpërmjet mekanizmit të reagimit negativ, bllokon përkthimin e ALA - sintazës dhe për rrjedhojë zvogëlohet sinteza e ALA dhe PBG, me çka arrihet lehtësimi i simptomave.
Eritropoetike kongjenitale porfiria është një çrregullim kongjenital edhe më i rrallë i trashëguar në mënyrë autosomale recesive. Natyra molekulare e kësaj sëmundjeje nuk dihet saktësisht; megjithatë, u zbulua se karakterizohet nga një çekuilibër i caktuar në aktivitetet relative të uroporfirinogjen-III-kosintazës dhe uroporfirinogjen-1-sintazës. Formimi i uroporfirinogjenit I tejkalon në mënyrë sasiore në mënyrë të konsiderueshme sintezën e izomerit normal të uroporfirinogjenit III në rrugën e sintezës së hemit. Megjithëse çrregullimi gjenetik shtrihet në të gjitha qelizat, ai manifestohet, për arsye të panjohura, kryesisht në indet eritropoetike. Pacientët me porfiri eritropoetike kongjenitale ekskretojnë sasi të mëdha izomerë të tipitUnë uroporfirinogjen dhe koproporfirinogjen; në urinë, të dyja këto komponime oksidohen në mënyrë spontane në pigmente fluoreshente të kuqe uroporfirin I dhe koproporfirinë I. U raportua se kishte një rritje të lehtë në përqendrimin e uroporfirinës III, por raporti i izomerëve të tipit I dhe III ishte afërsisht 100: 1. Qelizat e kuqe të gjakut në qarkullim përmbajnë një sasi të madhe të uroporfirinës 1, megjithatë, përqendrimi më i lartë i kësaj porfirina u vu re në qelizat e palcës kockore (por jo në hepatocitet).
Është shënuar fotosensitiviteti i lëkurës, për shkak të natyrës së spektrit të përthithjes së përbërjeve të porfirinës, të cilat formohen në sasi të mëdha. Në pacientët, vërehen çarje në lëkurë, shpesh vërehen fenomene hemolitike.
Koproporfiria trashëgimore -Çrregullim autozomal dominues për shkak të mungesës genoxidase koproporfike- një enzimë mitokondriale përgjegjëse për shndërrimin e koproporfirinogjenit III në protoporfirinogjen IX. Koproporfirinogjeni III ekskretohet në sasi të mëdha nga trupi me feces dhe për shkak të tretshmërisë së tij në ujë, ekskretohet në sasi të mëdha në urinë. Ashtu si uroporfirinogjeni, koproporfirinogjeni oksidohet me shpejtësi në dritë dhe ajër, duke u kthyer në pigmentin e kuq, koproporfirinë.
Aftësia e kufizuar e kësaj sëmundjeje për të sintetizuar hemin (veçanërisht në kushte stresi) çon në depresion të ALA-siitazës. Si rezultat, ka një formim të tepruar të ALA dhe porfobilinogjenit, si dhe ndërmjetësve të tjerë në rrugën e sintezës së temës, të formuar në fazat që paraprijnë fazën e bllokuar trashëgimore. Prandaj, në pacientët me koproporfiri trashëgimore, gjenden të gjitha shenjat dhe simptomat që lidhen me një tepricë të ALA dhe porfobilinogjenit, të cilat janë karakteristike për porfirinë akute intermitente, por përveç kësaj ato kanë një rritje të ndjeshmërisë ndaj fotos për shkak të pranisë së sasive të tepërta të koproporfirinogjeneve dhe uroporfirinogjenet. Në këtë sëmundje, administrimi i hematinës gjithashtu mund të shkaktojë të paktën shtypjen e pjesshme të ALA sintazës dhe përmirësimin e simptomave të shkaktuara nga mbiprodhimi i ndërmjetësve biosintetikë të hemit.
Porfiria e mozaikut , ose fotoporfiria trashëgimore, është një çrregullim autosomik dominant në të cilin ka një bllokim të pjesshëm të shndërrimit enzimatik të protoporfirinogjenit në hem. Normalisht, ky transformim kryhet nga dy enzima, protoporfirinogjen oksidaza dhe ferrokelataza, të lokalizuara. në mitokondri. Duke gjykuar nga të dhënat e marra për kulturën e fibroblasteve të lëkurës, në pacientët me mozaik porfirie, përmbajtja e protoporfirinogjen oksidazës është vetëm gjysma e sasisë normale. Në pacientët me mozaik porfirie, ekziston një mangësi relative në përmbajtjen e hemit në kushte stresi, si dhe një gjendje e depresuar e sintazës hepatike ALA. Siç u përmend më lart, aktiviteti i shtuar i ALA sintazës çon në mbiprodhimin e të gjithë ndërmjetësve të sintezës së hemit në vendet përpara fazës së bllokimit. Kështu, pacientët me mozaik porfirie ekskretojnë sasi të tepërta të ALA, porfobilinogjenit, uroporfirinës dhe koproporfirinës në urinë, dhe uroporfirinës, koproporfirinës dhe protoporfirinës në feces. Urina e pacientëve është e pigmentuar dhe fluoreshente, dhe lëkura është e ndjeshme ndaj dritës në të njëjtën mënyrë si te pacientët me porfiri të vonuar të lëkurës (shih më poshtë).
Porfiria e vonshme e lëkurës , është ndoshta forma më e zakonshme e porfirisë. Zakonisht shoqërohet me një lloj dëmtimi të mëlçisë, veçanërisht me konsumimin e tepërt të alkoolit ose mbingarkesën me jone hekuri. Natyra e çrregullimit metabolik nuk është përcaktuar saktësisht, por shkaku i mundshëm është një mungesë e pjesshme e uroporfirinogjen dekarboksilazës. Çrregullimi duket se transmetohet si një tipar autosomik dominant, por penetrenca gjenetike është e ndryshueshme dhe në shumicën e rasteve varet nga prania e mosfunksionimeve të mëlçisë. Në përputhje me parashikimet, urina përmban sasi të shtuara të uroporfirinave të tipit I dhe III; në të njëjtën kohë, sekretimi urinar i ALA dhe porfobilinogjenit është relativisht i rrallë. Ndonjëherë urina përmban një sasi shumë të konsiderueshme porfirine, duke i dhënë asaj një nuancë rozë; kur acidifikohet, më shpesh jep fluoreshencë rozë në rajonin ultravjollcë.
Mëlçia përmban sasi të mëdha porfirinash dhe për këtë arsye fluorescon fuqishëm, ndërsa eritrocitet dhe qelizat e palcës kockore nuk kanë fluoreshencë. Manifestimi kryesor klinik në porfirinë lëkurore tardive është i rritur fotosensitiviteti i lëkurës. Në pacientët, nuk ka as rritje të aktivitetit të ALA sintazës, as, në përputhje me rrethanat, përmbajtje të tepërt në urinë të porfobilinogjenit dhe ALA; kjo lidhet me mungesën e sulmeve akute karakteristike për porfirinë akute intermitente.
Protoporfiria , ose protoporfiria eritropoetike duket se është për shkak të mungesës së trashëguar kryesisht ferrokelataza në mitokondritë e të gjitha indeve; klinikisht kjo sëmundje shfaqet si urtikarie akute e shkaktuar nga ekspozimi ndaj rrezeve të diellit. Eritrocitet, plazma dhe feçet përmbajnë sasi të larta të protoporfirinës IX, dhe retikulocitet (qelizat e kuqe të papjekura të gjakut) dhe lëkura (kur ekzaminohet me biopsi) shpesh fluoreshojnë me dritë të kuqe. Mëlçia ndoshta gjithashtu kontribuon në rritjen e formimit të protoporfirinës IX, por nuk vërehet ekskretimi urinar i porfirinave dhe pararendësve të tyre.
Sinteza e hemoglobinës
Hemi i sintetizuar në mitokondri induktohet nga sinteza e zinxhirëve të globinës në poliribozome. Gjenet e zinxhirit të globinës janë të vendosura në kromozomet 11 dhe 16.
Zinxhirët e globinës formojnë globula dhe lidhen me hemin. 4 globula janë të kombinuara në mënyrë jokovalente për të formuar hemoglobinë.
Hemoglobina fillon të sintetizohet në fazën e eritroblasteve bazofile dhe përfundon në retikulocite. Në retikulocitet bëhet edhe sinteza e purinave, pirimidinave, fosfatideve, lipideve. Një tregues i ndjeshëm biokimik për dallimin e retikulociteve nga qelizat e pjekura është humbja e glutaminazës nga këto të fundit. Glutamina në retikulocitet është një burim karboni për sintezën e porfirinës dhe azoti për sintezën e purinës.
Struktura e hemoglobinës
Hemoglobina - kromoproteina tetramerike, ka një masë prej 64500 Da, përbëhet nga 4 heme dhe 4 globina. Globinat përfaqësohen nga vargje polipeptide të llojeve të ndryshme,,, , etj.-zinxhiri përmban 141 AA, dhe zinxhiri - 146 AA. Seksione të veçanta të zinxhirëve polipeptidë formojnë spirale me dorën e djathtë, një rregullim i veçantë në hapësirën e të cilave formon rruaza. Globula -nënnjësitë përmbajnë 8-helika, a-nënnjësitë –7. Hemi ndodhet në boshllëqet midis bobinave E dhe F të globinës, duke u ngjitur përmes histidinës F 8 në spiralen F me anë të lidhjes 5 koordinuese të hekurit. Mbetjet hidrofobike të aminoacideve që rrethojnë hemin parandalojnë oksidimin e hekurit nga uji. 4 globula me pjesëmarrjen e lidhjeve hidrofobike, jonike dhe hidrogjenore formojnë një tetramer sferik të hemoglobinës. Lidhjet më të forta, kryesisht për shkak të lidhjeve hidrofobike, formohen midis-dhe-globulave. Si rezultat, formohen 2 dimerë 1 1 dhe 2 2. Dimerët lidhen me njëri-tjetrin kryesisht me lidhje polare (jonike dhe hidrogjenore), prandaj bashkëveprimi i dimerëve varet nga pH. Dimerët lëvizin lehtësisht në lidhje me njëri-tjetrin. Në qendër të tetramerit, globulat janë lirshëm ngjitur me njëra-tjetrën, duke formuar një zgavër.
Funksionet e hemoglobinës
Siguroni transferimin e oksigjenit nga mushkëritë në inde. Rreth 600 litra në ditë;
Merr pjesë në transferimin e dioksidit të karbonit dhe protoneve nga indet në mushkëri;
Rregullon numërimin e gjakut.
Çdo patologji në metabolizmin e hekurit shoqërohet me zhvillim aneminë – një gjendje e dhimbshme e karakterizuar ose nga një ulje e numrit të qelizave të kuqe të gjakut ose nga një rënie në kontributin e hemoglobinës. Kjo është një nga sëmundjet më të zakonshme. Ndodh si rezultat i një sërë arsyesh:
a) për shkak të mungesës së hekurit në dietë (tek vegjetarianët, gjatë agjërimit, kur përdorin dieta të ndryshme për humbje peshe, tek gratë shtatzëna, gjatë ushqyerjes me gji, tek adoleshentët me rritje të shpejtë);
b) për shkak të përthithjes së dëmtuar në traktin gastrointestinal (me hiposekretim të acidit klorhidrik, proteazave, pas gastrektomisë nëntotale, me ndryshime në ekuilibrin e lëndëve ushqyese - mungesë e askorbatit, suksinatit, një tepricë e acidit fitik, fibrave, me dëmtim të mukoza e zorrëve në sëmundjen e ulçerës peptike, hernie diafragmatike, kolit ulceroz, pas trajtimit me salicilate, steroide, me helminthiasis, veçanërisht me lezione me krimb kamxhik, krimb gjilpërash);
c) për shkak të rezervave të pamjaftueshme të hekurit;
d) për shkak të ndryshimeve në lidhjet individuale të metabolizmit të këtij metali kalimtar (me frenim të aktivitetit të enzimave të sintezës së hemit);
e) pas një çlirimi të shtuar të joneve të këtij metali nga trupi (me humbje akute dhe kronike të gjakut, pas menstruacioneve të rënda - polimenorre, me hemorroide, ulçera të ndryshme në stomak, zorrë, pas episodeve të përsëritura të hemoptizës).
Shenjat kryesore klinike të anemisë: dobësi, palpitacione, lodhje, mungesë mendjeje, zbehje, gulçim.
Në varësi të shkallës së ruajtjes së sasisë së hekurit në organizëm, sekretohen anemitë me mungesë Fe, Fe të mjaftueshme, Fe të tepërt. Rreth 98 - 99% e të gjitha rasteve të sëmundjeve të tilla ndodhin në opsionin e parë. Të tjerat bazohen në anomalitë në përdorimin e hekurit në sintezën e hemit. Jonet e metalit kalimtar që nuk përfshihen në këtë përbërje fillojnë të depozitohen në formë hemosiderinë (të trashëguara dhe të fituara hemokromatoza ) në organe dhe inde (mëlçi, pankreas, miokard, nyje, lëkurë) me shtypjen e mëvonshme të funksioneve të tyre. Treshja e mëposhtme e shenjave mund të gjurmohet: cirroza e mëlçisë, diabeti mellitus, ngjyrimi në bronz i lëkurës (diabeti prej bronzi). Dhe meqenëse simptomat e anemisë zhvillohen paralelisht (për shkak të mungesës së hemoglobinës heme në eritrocite), është veçanërisht e rrezikshme përdorimi i preparateve të hekurit si agjentë terapeutikë.
Një shembull i anemive të tilla është aciduria metilmalonike , e cila bazohet në dëmtimin gjenetik të punës së enzimës që përmban B12 - metilmalonil-CoA mutaza , përgjegjës për izomerizimin e metilmalonil-Co A në suksinil-CoA - një nga substratet në gjenezën e hemit.
Patogjeneza Sëmundja e Heilmeyer vjen në faktin se funksioni i gjenit përgjegjës për sintezën e transferrinës është i bllokuar. Në mungesë të tij, sistemi i lëshimit të hekurit në palcën e eshtrave nuk funksionon, në këtë drejtim, formimi i hemit shtypet, zhvillohet anemia. Sëmundja trashëgohet në mënyrë autosomale recesive.
Emërtohen sëmundjet e bazuara në dëmtimin në sintezën e hemit porfiritë ... Në varësi të lokalizimit të çrregullimeve, dallohen llojet eritropoetike (dëmtime në metabolizmin e porfirinave në palcën e eshtrave) dhe hepatike (ndryshime të ngjashme në hepatocite). Më shpesh kjo është për shkak të gjenetikës, më rrallë është fituar në natyrë. Aktualisht, blloqet e të gjitha enzimave që marrin pjesë në sintezën e hemit janë regjistruar.
Porfiria (më saktë, forma e saj eritropoetike e trashëguar) u përshkrua për herë të parë nga Schultz (1874) dhe Gunther (1911). Megjithatë, kronikat historike të mesjetës kanë ruajtur përshkrime të familjeve, anëtarët e të cilave shfaqnin tipare karakteristike të formave të rënda të kësaj vuajtjeje, të manifestuara me simptoma të lëkurës, neurologjike dhe abdominale (simptoma abdominale akute, kriza epileptike, polineuriti, halucinacione, verbëri), si. si dhe një çlirim anormalisht i lartë i porfirinave me urinë ose feces. Disa shenja të sëmundjes janë një nuancë e kuqe e dhëmbëve dhe eshtrave, një ngjyrë e veçantë e lëkurës, e ndryshuar nga flluska, ulçera dhe plagë; stili i jetesës së natës i shkaktuar nga fotodermatiti, shkëlqimi spontan i disa indeve dhe sekrecioneve të pacientit, tekat e shijes që lidhen me aneminë janë aq të gjalla dhe të pazakonta sa të kujtojnë përshkrimet e pamjes dhe sjelljes së vampirëve mitikë.
Porfiritë e fituara janë më të rralla, shkaktarët e të cilave janë helmimi me kripëra të metaleve të rënda, të cilat, duke ndërvepruar me grupet sulfhidrile të sintazës aminolevulinate ose ferrokelatazës, shtypin aktivitetin e tyre në gjenezën e hemit. Si rezultat, protoporfirina grumbullohet në eritrocite, përmbajtja e hekurit rritet në plazmën e gjakut, depozitohet në organe dhe inde, duke provokuar formimin e hemosiderozës.
Natyrisht, patologjia e sintezës së globinës është e trashëguar. Ekzistojnë dy forma kryesore të çrregullimeve: dëmtimi i një aminoacidi të vetëm në strukturën e proteinave ( hemoglobinoza ) dhe shtypja e prodhimit të çdo zinxhiri polipeptid të globinës ( talasemitë ).
Aktualisht, janë përshkruar më shumë se 300 lloje të hemoglobinave patologjike. Hemoproteinat e para të modifikuara u emëruan duke përdorur shkronja latine (C, D, E, M, S), por kur numri i Hb të modifikuar tejkaloi numrin e shkronjave në alfabet, ato filluan të emërtohen sipas vendit të zbulimit (Kansas, Boston, San Yose, Hiroshima, Richmond, etj.).
Natyra heterogjene e dëmtimit gjenetik (zëvendësimi, futja, zhvendosja e kornizës, zgjatja e zinxhirit etj.) çon në pasoja të ndryshme (ndryshon afiniteti për oksigjenin, ulet qëndrueshmëria e eritrociteve, gjë që manifestohet me rritje të hemolizës, cianozë). Kjo përcaktohet jo vetëm nga ashpërsia e zhvendosjeve në sekuencën nukleotide, por edhe nga natyra e aminoacideve të ndryshuara. Nëse analogët zëvendësohen, për shembull, glutamati për aspartat, atëherë ky opsion nuk manifestohet në asnjë mënyrë. Praktikisht të shëndetshëm janë edhe bartësit e Hb San Jose, në të cilin në pozicionin e 7-të të vargut beta, glutamati zëvendësohet nga glicina, e cila ndikon vetëm në lëvizshmërinë elektroforetike të kësaj proteine.
Nëse struktura e vendit të mutuar ndryshon befas, atëherë gjasat për të zhvilluar shenja serioze klinike janë të larta. Veçanërisht të rrezikshme janë lezionet e vendosura në zonat e kontaktit (vendet ku nënnjësitë individuale lidhen me tetramerët) ose në xhepat ku ndodhet hemi. Në këto raste prishet kompleksizimi i tetramerit heterogjen, gjë që ndikon në qëndrueshmërinë e embrionit dhe rrit mundësinë e abortit. Një shembull është Hb Philly, në të cilin fenilalanina zëvendëson tirozinën, e cila në një proteinë normale formon një lidhje hidrogjeni me nënnjësi të tjera. Pas një mutacioni të tillë, ekzistenca e një tetrameri të ndryshuar gjenetikisht bëhet e pamundur - micela bie. Boston Hb karakterizohet nga zëvendësimi i tirozinës për histidinën në pozicionin 58 të zinxhirit alfa. Histidina, nga ana tjetër, zakonisht formon një lidhje koordinimi me hekurin hem dhe tirozina oksidon jonin në fenolate të hekurit; methemoglobina që rezulton provokon hipoksi.
Në hemoglobinën Genova, për shkak të humbjes së valinës, fragmenti zhvendoset, pjesa e mbetur e glutamatit ndodhet brenda micelës, vendi deformohet, gjë që redukton afinitetin për oksigjenin dhe zhvillohet cianoza.
Pasojat më të favorshme janë të mundshme nëse mutacionet prekin aminoacidet që krijojnë sipërfaqen e micelës. Një shembull klasik është Hb S, në pozicionin e 6-të të zinxhirit beta, glutamati i të cilit zëvendësohet nga valina, domethënë një përbërje acidike zëvendësohet nga një hidrofob. Dhe meqenëse dëmtimi është në sipërfaqe, një ndryshim i tillë zvogëlon ngarkesën dhe tretshmërinë e hemoglobinës, dhe molekulat e saj individuale, duke u përplasur, ngjiten së bashku për shkak të formimit të ndërveprimeve hidrofobike të valinave të micelave të ndryshme. Një grumbullim i tillë zgjat filamentet e hemoproteinës, gjë që redukton më tej tretshmërinë. Kjo dukuri ndikon në formën e rruazave të kuqe të gjakut: ato marrin formën e një drapëri (Sickle cell - sickle cell). Prandaj sëmundja quhet anemi në formë drapëri, ose hemoglobinoza S . Membrana e dëmtuar e eritrociteve është më pak e qëndrueshme, gjë që provokon hemolizë, trombozë. Pacientët karakterizohen nga kriza hemolitike me sindromë dhimbjeje akute, simptoma të dëmtimit të mëlçisë, verdhëz intensive dhe ndoshta formimin e gurëve në traktin biliar. Frekuenca e pranisë së një hemoproteine të tillë në Shtetet e Bashkuara është 8-9% (në afrikano-amerikanët), dhe në disa rajone të Greqisë arrin në 40%.
Talasemitë(thalassa - det) - sëmundje trashëgimore, të cilat, siç u përmend më lart, bazohen në bllokun e sintezës së të gjithë zinxhirit të globinës. Në këtë rast, micelat sintetizohen në qelizat eritroide, që shpesh përfaqësojnë tetramerë homogjenë, për shembull, të përbërë vetëm nga zinxhirë beta ose gama. Hemoglobina H, e përbërë nga 4 nënnjësi beta, është në gjendje vetëm të lidhë oksigjenin, por jo të japë. Me frenimin e sintezës së zinxhirëve alfa, embrioni homozigot nuk është i zbatueshëm, një abort ndodh në shtatzëninë e hershme. Fëmija me beta talasemia (Sëmundja Cooly) (një bllok i gjenezës së zinxhirit beta) lind praktikisht i shëndetshëm (në fund të fundit, hemoglobina, duke përfshirë zinxhirët alfa dhe gama, është e nevojshme për rritjen dhe zhvillimin e fetusit). Çdo muaj fillojnë të shfaqen shenja patologjike, të përcaktuara nga shkalla e hipoksisë, rritja e peroksidimit të lipideve, dëmtimi i membranave të eritrociteve (anemia e rëndë hemolitike, vazhdimësia e përmbajtjes së lartë të HbF (deri në 20-30%), zhvillimi fizik dhe mendor i dëmtuar, fëmija refuzon të marrë gji, fytyra fiton tipare mongoloide - mollëzat dalin përpara, baza e urës së hundës shtypet, hunda rrafshohet). Për një moshë më të madhe janë karakteristike krizat hemolitike, një gjendje febrile dhe mund të zhvillohet insuficienca kardiake.
Përhapja e këtyre sëmundjeve ndryshon sipas rajonit. Ato janë veçanërisht të zakonshme në brigjet e deteve (Mesdheu, i Zi). Në Transkaukazi, deri në 10% të bartësve të gjeneve të tilla janë regjistruar.
DISHEMOGLOBINEMIA
Gjendjet patologjike janë relativisht të zakonshme, të cilat bazohen në çekuilibër në forma të ndryshme të hemoglobinës. Në eritrocitet e një të rrituri të shëndetshëm, niveli methemoglobina nuk kalon 2% të përmbajtjes totale. Megjithatë, nën ndikimin e oksideve të ndryshme të azotit, nitrateve inorganike, komponimeve organike nitro (amil nitrit, nitrobenzen, nitrofenol, trinitrotoluen, nitroaniline), derivatet amino (hidroksilaminë, fenilhidrazinë, aminofenole, acid p-aminobenzoik, klorate, aniline), permanganate, pirone disa barna (nitroglicerina, anestezina, furadonin, barbituratet, aspirinë, etj.), Bojërat me aftësi oksiduese - përqendrimi i saj rritet ndjeshëm ( met-hemoglobinemia ). Si rezultat, funksioni kryesor i hemoglobinës prishet - transferimi i oksigjenit nga mushkëritë në inde bllokohet dhe zhvillohet hipoksi.
Të gjithë formuesit e methemoglobinës, duke ulur rezistencën osmotike të eritrociteve, përshpejtojnë hemolizën e tyre. Gjatë oksidimit të Hb në methemoglobinë, gjenerohen radikale aktive të oksigjenit, të cilat mund të marrin pjesë në proceset e dëmtimit oksidativ të eritrociteve. LPO aktivizohet, metabolizmi lipidik i membranave të qelizave të kuqe të gjakut është i shqetësuar, ekuilibri në sistemin LPO - AOD është zhvendosur. Simptoma kryesore është cianoza; nëse përmbajtja e methemoglobinës tejkalon 30%, shfaqen dobësi, marramendje, takikardi, dhimbje koke; kur akumulohet deri në 50%, zhvillohet insuficienca kardiovaskulare. Rreziku i helmimit nga nitritet rritet me përdorimin e perimeve, salsiçeve, mishit të zier dhe ujit të pijshëm me cilësi të dobët. Janë përshkruar gjithashtu raste të methemoglobinemisë trashëgimore (në bartës të Hb Boston) (shih më lart).
Grupi i helmeve të gjakut që formojnë pigmente patologjike përfshin oksid karboni (oksid karboni). Duke depërtuar në trup, CO absorbohet nga eritrocitet, ndërvepron me hemoglobinën e hekurit, duke formuar një përbërje mjaft të qëndrueshme - karboksihemoglobina , vlera e së cilës te joduhanpirësit nuk kalon 0,25% të sasisë totale të proteinës bazë të eritrociteve. Në gjakun e duhanpirësve, numri i tij rritet në 6-7%. Në një presion të pjesshëm më të lartë të monoksidit të karbonit ( karboksihemoglobinemia ) frenohet oksigjenimi i hemoproteinës, zhvillohet hipoksia. Përveç kësaj, CO ka aftësinë të bie në kontakt me proteinat e tjera që përmbajnë hem (mioglobina, citokromet, peroksidaza, katalaza), duke prishur funksionet e tyre.